Current insights into inherited bone marrow failure syndromes
Article information
Abstract
Inherited bone marrow failure syndrome (IBMFS) encompasses a heterogeneous and complex group of genetic disorders characterized by physical malformations, insufficient blood cell production, and increased risk of malignancies. They often have substantial phenotype overlap, and therefore, genotyping is often a critical means of establishing a diagnosis. Current advances in the field of IBMFSs have identified multiple genes associated with IBMFSs and their pathways: genes involved in ribosome biogenesis, such as those associated with Diamond-Blackfan anemia and Shwachman-Diamond syndrome; genes involved in telomere maintenance, such as dyskeratosis congenita genes; genes encoding neutrophil elastase or neutrophil adhesion and mobility associated with severe congenital neutropenia; and genes involved in DNA recombination repair, such as those associated with Fanconi anemia. Early and adequate genetic diagnosis is required for proper management and follow-up in clinical practice. Recent advances using new molecular technologies, including next generation sequencing (NGS), have helped identify new candidate genes associated with the development of bone marrow failure. Targeted NGS using panels of large numbers of genes is rapidly gaining potential for use as a cost-effective diagnostic tool for the identification of mutations in newly diagnosed patients. In this review, we have described recent insights into IBMFS and how they are advancing our understanding of the disease's pathophysiology; we have also discussed the possible implications they will have in clinical practice for Korean patients.
Introduction
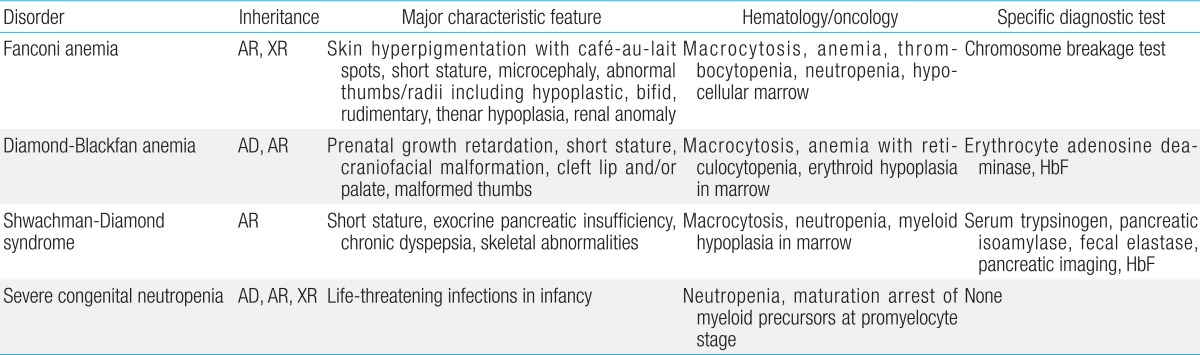
Inherited bone marrow failure syndrome (IBMFS) encompasses a heterogeneous and complex group of genetic disorders characterized by physical malformations and insufficient blood cell production. Bone marrow failure (BMF) manifests symptoms similar to aplastic anemia, mono- or bicytopenias, depending on the affected cell lineage. IBMFS includes disorders like Fanconi anemia (FA), dyskeratosis congenita (DC), Diamond-Blackfan anemia (DBA), severe congenital neutropenia (SCN), Shwachman-Diamond syndrome (SBDS), and thrombocytopenia with absent radii. Various features of each of these disorders are summarized in Table 1. There is often substantial phenotypic overlap between these disorders, making genotyping a critical means for establishing a diagnosis. Mutations in many different genes have been associated with the development of BMF; these findings have enhanced our understanding of normal hematopoiesis and its disruption in IBMFs patients.
Inherited bone marrow failure syndrome
The genetic etiology of IBMFS includes germline mutations in several key biological processes, such as DNA repair, telomere biology, and ribosomal biogenesis, which underlie FA, DC, and DBA, respectively. Additionally, as these disorders are associated with developmental abnormalities and an increased risk of cancer, early and adequate genetic diagnosis is required for proper management and follow-up. Furthermore, the heritable nature of these conditions and the anticipated risk for other family members and future generations underscore the value of this information for genetic counseling. Recent advances using new technologies such as next generation sequencing (NGS) have identified new candidate genes associated with the development of BMF.
In this review, we have described recent insights into IBMFS and shown how they are advancing our understanding of disease pathophysiology; we have also discussed their possible implications in clinical practice for Korean patients.
Fanconi anemia
Fanconi anemia (FA, MIM #227650) is a complex disease that can affect many systems in the body. FA is characterized by congenital abnormalities, progressive development of BMF, and an increased susceptibility to malignancy, including myelodysplastic syndrome (MDS), leukemia (especially acute myeloid leukemia [AML]) and solid tumors (particularly squamous cell carcinoma). Affected individuals often (but not always) present with physical abnormalities, including short stature, Fanconi facies and microphthalmia, thumb and radius deformities, skin pigmentation such as café-au-lait spots, and cardiac, renal, genitourinary, and/or other malformations1). Approximately 30% of patients with FA have no overt somatic abnormalities2). The majority of patients present symptoms toward the end of their first decade of life. However, increasingly, patients are being diagnosed in adulthood and many patients diagnosed in childhood are surviving into adulthood3). Increased fetal hemoglobin, serum alpha-fetoprotein, and macrocytosis are commonly observed in FA, but their absence does not rule out the possibility of the disease. A precise diagnosis can be made on the basis of a careful family-history survey, physical examination, and a positive chromosomal breakage test in the majority of cases. Cells from FA patients display a high frequency of spontaneous chromosomal breakage and hypersensitivity to DNA cross-linking agents such as mitomycin C and diepoxybutane4). Chromosomal instability, especially upon exposure to an alkylating agent, is present in affected patients and is the basis for a diagnostic test (Fig. 1). However, inconclusive chromosome breakage results caused by somatic mosaicism may complicate the diagnosis of FA. If the test is negative in peripheral blood lymphocytes and yet the clinical setting is highly suspicious, skin fibroblasts must be assessed5,6).

Karyotype with induction of chromosome breakage by using mitomycin C, in patients diagnosed with Fanconi anemia (arrows: chromosomes with breaks, fusions, and radials).
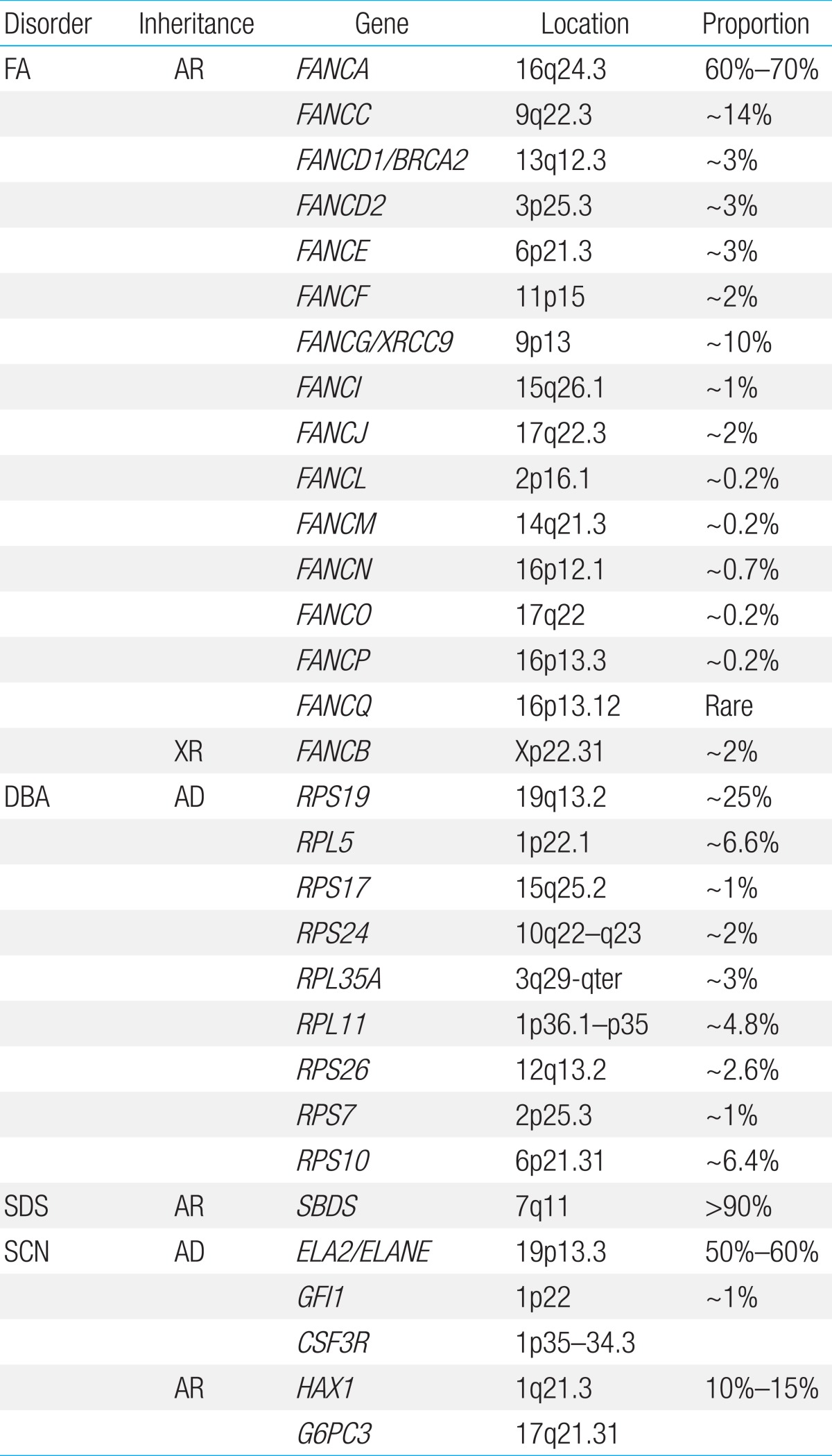
FA is a genetically heterogeneous disease. With the exception of X-linked recessive FA-B, FA-associated genes are inherited in an autosomal recessive pattern. At least 16 genes exist that are responsible for the known FA complementation groups (Table 2)7,8). All FA products function in a common DNA repair signaling pathway that closely cooperates with other DNA repair proteins for resolving interstrand cross-links during replication9). FA-A, B, C, E, F, G, L, and M associate in a nuclear core complex, which is required for FANCD2 activation via monoubiquitination during the S phase and in response to DNA damage10). Germline mutations in any of the FA genes result in markedly reduced or abolished protein function and deficient DNA repair. Among them, FANCA (60%-70%), FANCC (up to 14%), and FANCG (up to 10%) mutations accounted for more than 90% of FA in a Western population11). Concerning Asian populations, genetic abnormalities in FA have been studied in a Japanese population. FANCA and FANCG mutations have been detected in 70%-80% and 10%-22%, respectively, of Japanese FA patients12). Recently, investigation of genetic abnormalities revealed that mutations in FANCA and FANCG are common in Korean FA patients (46% and 54%, respectively)13,14). A recent report also identified four common founder mutations, including two FANCA mutations (c.2546delC and c.3720_3724delAAACA) and two FANCG mutations (c.307+1G>C and c.1066C>T), which had previously been shown to be common in an East Asian FA population13). Based on results from previous research on Korean and Japanese patients, our suggestion is a mutation screening workflow for East Asian FA patients (Fig. 2).
Genes associated with inherited bone marrow failure syndromes
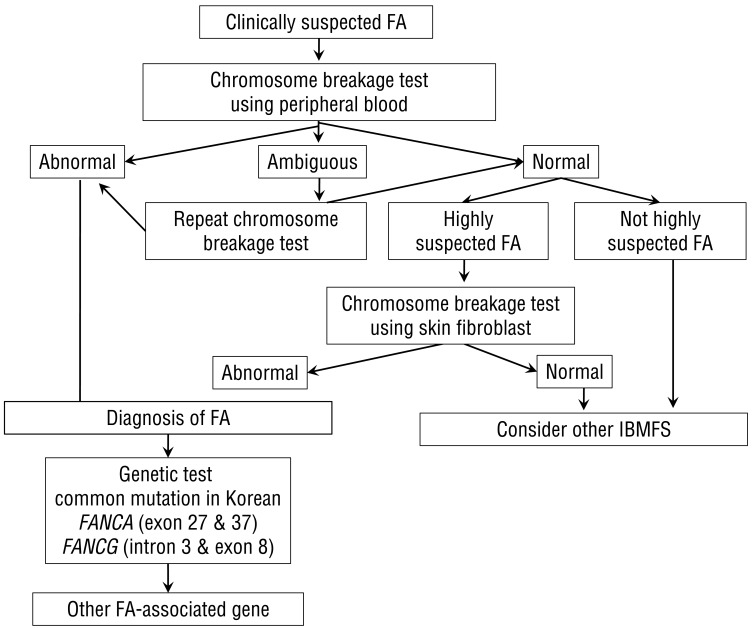
Diagnostic algorithm for Fanconi anemia (FA). IBMFS, inherited bone marrow failure syndrome.
Whole exome sequencing (WES) has accelerated the identification of mutations in rare cases of FA without a known genetic etiology. Targeted NGS has the potential to be used as a cost-effective diagnostic tool for the identification of FA-associated mutations in newly diagnosed patients. A study that sequenced germline DNA of 11 patients with known FA mutations on a custom NGS sequencing panel of eight FA genes (FANCA, FANCB, FANCC, FANCD1, FANCE, FANCG, FANCI, and FANCN) indicated the importance of NGS in uncovering somatic mosaicism by comparing different tissue types15). The combination of custom array comparative genomic hybridization (aCGH) to detect deletions and duplications of FA genes, WES to evaluate mutations, and RNA-Seq to evaluate gene expression changes resulting from these mutations proved to be an efficient means for the characterization of both the complementation group and the germline mutations16).
The most frequent malignancy in FA is AML, but solid tumors can develop at a very young age in patients17). A review of cases of FA reported in the literature showed that 9% of patients developed leukemia and 7% developed MDS, 5% had solid tumors, and 3% had liver tumors. The risk of developing solid tumors, particularly of the head and neck, skin, esophagus, and gynecologic areas also increases18). AML with complex hypodiploidy and loss of heterozygosity of 17p and congenital pigmented dermofibrosarcoma protuberans were reported in Korean FA patients19,20). Long-term follow-up of 12 FA patients helped track the development of AML during follow-up in two patients21). In addition, with the development of therapeutic techniques for FA such as hematopoietic stem cell transplantation and posttransplant care, more patients are surviving and being followed up for longer periods than before. With the prospect of a significantly extended lifespan, we must be prepared to treat problems related to nonhematological conditions that were previously considered secondary and to devise a follow-up plan for those patients. There is a critical need for nation-wide studies in order to assemble more genetic and clinical data to develop cost-effective diagnostic strategies that are optimal for Korean FA patients.
Diamond-Blackfan anemia
Diamond-Blackfan anemia (DBA, MIM# 105650) is an inherited congenital BMF syndrome characterized by hypoproliferative anemia, associated physical malformations, and a predisposition to cancer. The hallmark of classical DBA is normochromic macrocytic anemia with reticulocytopenia and the absence or insufficiency of erythroid precursors in an otherwise normocellular bone marrow (Fig. 3A)3). The disease is rare, with a reported incidence of 7 per million live births, and the anemia usually occurs in early infancy, with more than 90% of the patients being diagnosed before the age of 1 year22). Most DBA cases are sporadic, but the disease can be inherited with an autosomal dominant mode of inheritance. Although anemia is the most prominent feature, the disease is also associated with congenital malformations, mainly involving the head, upper limbs, heart, and the urogenital system, and growth retardation in approximately 50% of patients3). The clinical and hematologic manifestations in 56 Korean DBA patients were assessed in a previous report23). The annual incidence of DBA was found to be 6.6 cases per million and 74.1% were diagnosed before 1 year of age (median, 4 months). About 40% of patients showed congenital defects, including thumb deformities, ptosis, coarctation of the aorta, ventricular septal defect, and strabismus.
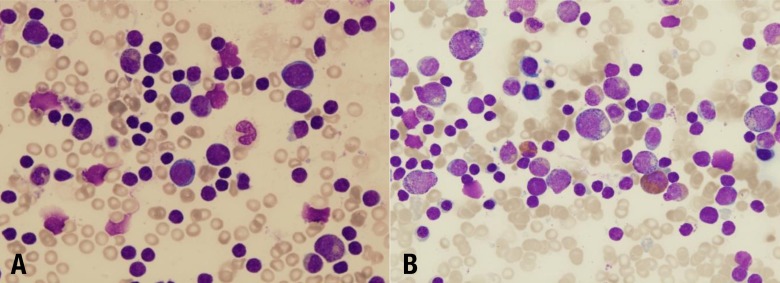
Bone marrow in Diamond-Blackfan anemia, revealing a severe lack of erythroid precursors (A: Wright's stain, ×400) and severe congenital neutropenia, showing increased promyelocytes and a marked absence of mature granulocytes (B: Wright's stain, ×400).
DBA has been associated with mutations and deletions in the large and small ribosomal protein (RP) genes, and genetic aberrations have been detected in approximately 50%-60% of patients. Since the identification of mutations in the RP S19 gene, which was the first DBA gene to be reported, mutations in an increasing number of genes encoding RPs of both the small (RPS) and large (RPL) ribosomal subunits have been reported. At present, 10 genes encoding RPs of the small (RPS24, RPS17, RPS19, RPS10, RPS26, and RPS7) and large (RPL35A, RPL5, RPL11, and RPL26) ribosomal subunits have been described in DBA patients24). In the Japanese cohort, 27% of the 45 DBA patients had mutations in the RP genes, and mutations in the RPS19 gene were detected in 11% of patients. Mutations in RPL5, RPL11, and RPS17 were also detected25). A recent study investigated the genetic abnormalities in nine Korean DBA patients and noted that seven distinct mutations, including three novel mutations, were identified in three RP genes, RPS19 (4/7), RPS26 (2/7), and RPS17 (1/7)26). Haploinsufficiency of RPs results in ribosome stress and activation of the TP53 tumor suppressor pathway, which is thought to be the main cause of clinical manifestations27). Decreased RPS19 mRNA expression and p53 overexpression were also seen in Korean DBA patients, supporting the idea that haploinsufficiency and p53 overactivation are the central pathways underlying the pathogenesis of DBA26).
Clinical suspicion and molecular genetic studies-even in patients without typical anomalies-are critically needed to better understand the genetic background and genotype-phenotype correlations of DBA and to optimize the treatment strategy28). A clear genotype-phenotype correlation was not apparent in patients with RPS19 mutations due to incomplete penetrance and a highly variable expression profile, even within the same family29). Additionally, much of our knowledge of the disease has been gained from patient registries in North America, United Kingdom, Italy, and France22,30,31). There have been only a few other genotype studies performed on Asian DBA patients. Among the DBA patients harboring RPS19 mutations, RPS19 alterations have not been correlated with any specific phenotype. When researchers compared the clinical features of the RPS19-mutated DBA patients in their cohort with those of the previously reported DBA patients who shared identical or associated genotypic aberrations, no apparent correlation was found within the RPS19-mutated patients. The whole genome approach involving WES and aCGH of the known DBA-associated genes encoding RPs has been successfully attempted and has revealed mutations in new DBA-associated genes32,33).
Long-term follow-up in the DBA registry identified that the rate of any solid tumor or leukemia was 5.4 folds higher in DBA patients than expected in a demographically matched comparison with the general population. MDS and AML have been reported in a few patients, suggesting an increased predisposition to hematologic malignancies. There are also cases in which the disease has evolved into aplastic anemia33). Lymphoma and osteosarcoma developed in two Korean DBA patients23). It is necessary to collect further data, including information regarding management pathways, from nationwide DBA registries, along with data on molecular analyses.
Shwachman-Diamond syndrome
Shwachman-Diamond syndrome (SDS, MIM #260400) is characterized by BMF, pancreatic insufficiency, and skeletal abnormalities. Features of pancreatic insufficiency are apparent early in infancy. The spectrum of hematologic abnormalities includes neutropenia, pancytopenia (~20%), MDS, and leukemia (~25%)34). Patients with SDS have an increased risk-as high as 30%-of developing MDS and/or AML by the age of 30 years35). Biallelic Shwachman-Bodian-Diamond protein (SBDS) gene mutations inherited in an autosomal recessive fashion are causally related to the disease36). SBDS plays an important role in the maturation of the 60S ribosomal subunit and, therefore, in ribosome biogenesis37). In over 90% of patients diagnosed with SDS, the most common mutations are c.258+2T>C, followed by c.183_184TA>CT36). We examined the characteristics of two unrelated Korean adolescent patients with diverse clinical manifestations of SDS, including short stature, neutropenia, thrombocytopenia, congenital lipomatosis of the pancreas, Legg-Calve-Perthes syndrome, congenital coxa vara, and osteoporosis. Direct sequencing of the SBDS gene revealed compound heterozygosity of two common mutations (c.[183_184TA>CT];[258+2T>C]) in one patient and homozygous mutations of c.[258+2T>C];[258+2T>C] in the other patient.
Prior to the advent of genetic testing, the diagnosis of SDS was based largely on the clinical criteria of neutropenia and exocrine pancreatic insufficiency. However, patients with SBDS gene mutations revealed an unexpectedly broad range of clinical phenotypes at presentation. The frequency of cryptic presentations of SDS raises the likelihood that SDS is an underdiagnosed disorder. Serum trypsinogen and pancreatic isoamylase were the most sensitive measures of clinically asymptomatic pancreatic dysfunction in SDS. Normal pancreatic imaging studies and normal fecal elastase do not rule out the diagnosis of SDS38). An early diagnosis of SDS is necessary for the early intervention and treatment of the many clinical symptoms of this disease and to predict the hematological prognosis of patients. In the present cases, however, a conclusive diagnosis seemed to be delayed even though the patients were being followed up continuously. Therefore, clinicians should take extra efforts to ensure rapid diagnosis and to carry out additional studies to determine the functions of SBDS-related proteins.
Severe congenital neutropenia
Neutropenia is defined as a reduction in the absolute number of neutrophils in the blood circulation below 1.5×109/L-1 in children over 1 year and below 2.0×109/L-1 in children aged between 2 and 12 months39,40). SCN, as its name indicates, is characterized by profound peripheral neutropenia (<0.5×109/L-1) for at least 3 months, whether intermittent or permanent. Patients with the congenital disorder usually present with recurrent, life-threatening infections in infancy. Patients who are diagnosed with SCN usually show maturation arrest in the early stages of neutrophil development in the bone marrow (Fig. 3B). In addition, SCN is commonly considered as a preleukemic syndrome, since the cumulative incidence for leukemia exceeds 25% after 20 years of observation. Leukemia occurs in both subtypes (autosomal dominant and recessive) of SCN41). Bone marrow examination usually reveals characteristic maturation arrest of myeloid precursors at the promyelocyte stage of differentiation.
SCN is inherited in autosomal dominant, autosomal recessive, X-linked, and sporadic patterns. Heterozygous neutrophil elastase gene (ELA2) mutations are the most frequent cause of SCN and are present in patients with sporadic and familial forms of SCN42,43). Autosomal recessive SCN is associated with biallelic mutations in the HAX1 gene and the G6PC3 gene44,45). Mutations in WAS are associated with X-linked inherited SCN46). Mutations in other genes (GFI1, CSF3R) are also known to be associated with SCN, demonstrating genetic heterogeneity47,48). Despite recent progress in gene discovery, an all-encompassing causal mechanism underlying SCN has not yet emerged. Genetic surveys in Korean SCN patients have been carried out leading to the identification of two mutations in the ELA2 gene (p.Ala57Val and p.Gly203Arg)49,50). However, we recognize that the genetic profiles in the majority of our SCN cases remain unresolved and require future investigations51).
Conclusions
The identification of the molecular actors and the insights they provide into the involved biological pathways, have helped us to understand the pathogenesis of IBMFS. Current advances in molecular technologies have come largely through the rapid identification of new gene mutations, which account for a far broader clinical spectrum. Clinical similarities between these syndromes may originate from the close association and similarity between the underlying molecular pathology of these disorders. Genetic advances have already found application in the clinic and have led to improved diagnosis, particularly for cases wherein the clinical manifestations are atypical. Accumulating data on the Korean population have revealed the prevalence of mutated genes in IBMFS, the types of mutations involved, and the challenge in identifying the clinical characteristics of patients. Early and correct disease recognition can make for more appropriate decisions leading to improved management and prevention of long-term sequelae of the disease. In addition, with the development of parallel testing of multiple BMF genes in a single sample using NGS, comprehensive genetic testing will be available, enabling the development of appropriate screening tools, discovery of new prognostic markers, and development more specific drugs.
These findings suggest ethnic differences in phenotypic expression, a feature that has been observed previously in other genetic diseases, including FA. It also highlights the need to define the clinical-genetic spectrum in different ethnic populations.
Acknowledgments
This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120175).
Notes
No potential conflicts of interest relevant to this article were reported.