A case of thanatophoric dysplasia type I with an R248C mutation in the FGFR3 gene
Article information
Abstract
Thanatophoric dysplasia (TD) is a short-limb neonatal dwarfism syndrome that is usually lethal in the perinatal period. It is characterized by shortening of the limbs, severely small thorax, large head with a prominent forehead, macrocephaly, curved femur, and flattened vertebral bodies. These malformations result from the mutation in fibroblast growth factor receptor 3 (FGFR-3) gene which is located on the short arm of chromosome 4. A definite diagnosis should be established by molecular genetic analysis to find out the abnormal mutations in the FGFR3 gene. We confirmed by detection of a R248C mutation in the FGFR3 gene in DNA analysis.
Introduction
Thanatophoric dysplasia (TD) is a lethal chondrodysplasia caused by fibroblast growth factor receptor 3 (FGFR3) gene mutation which is located on the short arm of chromosome 4. The most frequent mutations (p.R248C, p.S249C, p.Y373C) create a cysteine residue within the extracellular domain. It was first described in 1967 by Maroteaux and Lamy1). It is the most common form of lethal congenital skeletal dysplasia, with an incidence of 1 in 20,000 to 40,000 live births2). TD is divided into type I, characterized by micromelia with bowed femurs and type II, characterized by micromelia with straight femurs. Other features of type I and type II include: short limbs, large head with frontal bossing, depressed nasal bridge, narrow thoracic cage with severe respiratory insufficiency, flat vertebral body (platyspondyly), and short pelvis. To our knowledge, there have only been about 20 cases of clinically diagnosed TD reported in Korea. Most of them were diagnosed by prenatal ultrasound and all these pregnancies were terminated. There were a few neonatal cases that were diagnosed clinically and radiologically, but none of the cases was confirmed by DNA analysis. We report a case of thanatophoric dysplasia type I which was diagnosed by the detection of the Arg248Cys mutation in DNA analysis.
Case report
A male baby was born by Cesarean delivery due to breech presentation and respiratory distress at the gestational age of 36 weeks and 2 days.
The baby's mother was a 19-year-old primigravida, single mother and did not undergo any antenatal evaluation. She visited our hospital when her labor pains started. The prenatal two-dimensional ultrasound examination just before the delivery showed polyhydramnios and short-limbed dwarfism. She had no history of drug, alcohol, or tobacco abuse. The baby's parents were healthy, and there was no family history of congenital abnormalities. At birth, the baby's heart rate and body temperature were 52 beats/min and 36.7℃, respectively. His Apgar score was 1 and 1 at 1 and 5 minutes, respectively. General appearance was cyanotic blue colored. The findings of arterial blood gas analysis showed: pH 7.08, PaO2 44.7 mmHg, PaCO2 61.1 mmHg, HCO3 13.9 mmol/L and oxygen saturation was 75%, in spite of the oxygen therapy by face mask.
He was intubated and resuscitated immediately and then transferred to the neonatal intensive care unit. The infantogram showed a small thoracic cage and consistent with respiratory distress syndrome, including both lung fields with inadequate inspiration of white out. The surfactant replacement therapy was performed with mechanical ventilation in the assist-control mode. After that, the baby's respiratory and hemodynamic status had been stabilized.
The laboratory findings on admission day were WBC 24,760/mm3 (neutrophils 47%, lymphocytes 38%), a hemoglobin level of 17.7 g/dL, hematocrit 54.8%, platelet 239,000/mm3, serum Na 135 mmol/L, K 4.5 mmol/L, AST/ALT 59/14 IU/L, BUN/Cr 11.6/0.7 mg/dL, and CRP was non-reactive.
The body weight at birth was 2,740 g (25-50th percentile) and height was 39 cm (<5th percentile). The head circumference was 39.5 cm (>90th percentile), the thoracic circumference was 27 cm and the abdominal circumference was 30 cm. He had a large head with prominent forehead and midfacial hypoplasia, narrow thorax, very short limbs with a short neck, and a protuberant abdomen (Fig. 1).
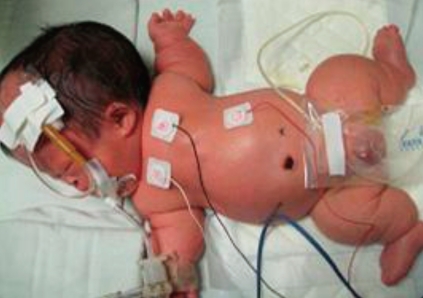
Full-body photograph acquired on the 2nd day of life, showing large head, markedly short limbs, and narrow thorax.
The radiographs showed a large calvaria with a small cranial base, marked thinning and flattening of the vertebral bodies, short ribs, hypoplasia of the pelvic bones, and small curved long bones so called telephone receiver femur (Fig. 2).
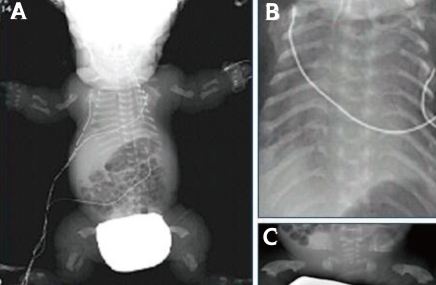
Newborn infantogram demonstrating thanatophoric dysplasia type I (TD1) findings, including platyspondyly, a severely hypoplastic pelvis, long bone shortness (A), hypoplasia of the lungs and thorax (B), and femoral bowing ("telephone receiver" femur) (C).
The baby was examined for R248C mutation by use of restriction digestion and direct sequencing. The results showed that the baby carried R248C mutation. In this case, nucleotide sequence analysis of cDNA revealed a C to T transition at nucleotide 742 (C742T) in one allele of the FGFR3 gene. This heterozygous 742C→T (R248C) mutation in the FGFR3 leading to an Arg248Cys (CGC→TGC) substitution (Fig. 3). This genetic condition causing type I thantophoric dysplasia. Despite of the mechanical ventilation, the baby's oxygen saturation levels are decreased day by day. Finally the baby died of respiratory failure, due to narrow thorax with pulmonary hypoplasia at 4 months of age.
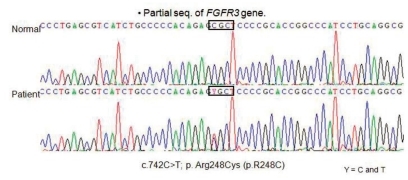
DNA sequencing of the FGFR3 gene showing Arg248Cys mutation in R248C.
Discussion
The term thanatophoric dysplasia (TD) originates from a Greek term for "death bearing". TD is characterized by an abnormal head, face, thorax and skeleton. The head anomaly consists of megacephaly, occasionally with a cloverleaf-shaped skull known as kleeblattschödel. In the face, excess skin usually yields a "boxers' face" appearance, with frontal bossing and a depressed nasal bridge. A hypoplastic thorax is common, which is disproportionately small in relation to the abdomen. Classically, a bell-shaped thorax has the shape of a champagne bottle cork and results in pulmonary hypoplasia. Ribs are hypoplastic, and the long bones of the extremities are short and curved, resulting in telephone receiver-shaped femurs. Vertebrae are flattened with diminution of the intervertebral spaces3-5).
Based on bone deformity pattern, TD has been divided into two types. Type I is more common and is characterized by curved long bones (with a 'telephone receiver' appearance) and severe platyspondylia, usually without a cloverleaf skull. In type II, the long bones are relatively straight, the platyspondylia is less severe, and a cloverleaf skull is usually present6). Affected individuals generally die within minutes or days after birth, usually from respiratory failure.
TD is an autosomal dominant disorder caused by sporadic de novo mutations in the FGFR3 gene mapped chromosome band 4p16.32). The FGFR3 gene is a cell surface protein consisting of extracellular, transmembrane and intracellular domains. It is particularly abundant in the cartilage growth plates7). Mutations in different domains of the FGFR3 gene are responsible for a variety of chondrodysplasias ranging from very mild hypochondroplasia to lethal TD including chondrodysplasia and Crouzon syndrome8). Essentially, all TD patients examined so far have had mutations in the transmembrane domains of FGFR3. TD II cases have a single recurrent mutation (A to G) in the tyrosine kinase domain of FGFR3, but TD I cases have different mutations affecting either the extracellular or intracellular domains of FGFR38). Although, a total of 12 distinct missense mutations have been described for TD I cases, the most common TD I mutation is a C to T transition (C742T) that leads to substitution of arginine by cysteine in 248 position of polypeptide (R248C) in the extracellular domain of FGFR39).
Molecular analysis of the FGFR3 gene using PCR-based methods is useful for the rapid screening of fetuses with sonographic findings suggestive of TD9). Early prenatal diagnosis of TD can be established by ultrasonography in the second trimester of gestation. However, it is not always possible to differentiate TD from other skeletal dysplasias by ultrasonography alone; thus, molecular genetic analysis of the fibroblast growth factor receptor 3 (FGFR3) gene is useful for the prenatal diagnosis of TD. Prenatal diagnosis can allow for elective abortion to be carried out, thereby avoiding possible complications such as later caesarean section, difficult vaginal delivery due to hydrocephalus, and malpresentation later in the pregnancy10).
The overall prognosis is poor. Most patients die of respiratory insufficiency due to reduced chest circumference and hypoplastic lung within 48 hours, although four to nine year survivals have been reported11, 12).
The first case of TD in Korea was reported in 198913), and there have been about 20 cases described since then. Most of the cases have been reported by obstetricians because they were diagnosed by the prenatal ultrasound and subsequently terminated.
However, this was the case of a single mother who had never had any prenatal evaluation until just before the delivery. The baby had typical TD I findings, such as a large head with a prominent forehead, a flat face, micromelia with telephone receiver femurs, and a narrow chest. Moreover, the Arg248Cys mutation in the extracellular region of the FGFR3 gene was identified with molecular study.
This case is the first report of a TD type I confirmed both radiologically and genetically in Korea.