Rhinovirus and childhood asthma: an update
Article information
Abstract
Asthma is recognized as a complex disease resulting from interactions between multiple genetic and environmental factors. Accumulating evidence suggests that respiratory viral infections in early life constitute a major environmental risk factor for the development of childhood asthma. Respiratory viral infections have also been recognized as the most common cause of asthma exacerbation. The advent of molecular diagnostics to detect respiratory viruses has provided new insights into the role of human rhinovirus (HRV) infections in the pathogenesis of asthma. However, it is still unclear whether HRV infections cause asthma or if wheezing with HRV infection is simply a predictor of childhood asthma. Recent clinical and experimental studies have identified plausible pathways by which HRV infection could cause asthma, particularly in a susceptible host, and exacerbate disease. Airway epithelial cells, the primary site of infection and replication of HRV, play a key role in these processes. Details regarding the role of genetic factors, including ORMDL3, are beginning to emerge. This review discusses recent clinical and experimental evidence for the role of HRV infection in the development and exacerbation of childhood asthma and the potential underlying mechanisms that have been proposed.
Introduction
Asthma is recognized as a complex disease resulting from interactions between multiple genetic and environmental factors. Cumulative evidence suggests that respiratory viral infections in early life constitute a major environmental risk factor for the development of asthma123). Previously, most research in this field focused on respiratory syncytial virus (RSV) infection45678). However, application of molecular diagnostic techniques to detect respiratory viruses has led to an increased awareness of the role of human rhinovirus (HRV) in wheezing illnesses during infancy and childhood asthma. Furthermore, recent large birth cohort studies have even suggested that HRV infection in the first years of life could be a much stronger risk factor for the subsequent development of asthma than RSV infection, at least in high-risk populations9). HRVs are also the most common viral pathogens isolated from children and adults with asthma exacerbations. Nonetheless, the fundamental mechanism that underlies these associations is still largely unknown. This review discusses the recent clinical and experimental evidence for the role of HRV infection in the development and exacerbation of childhood asthma and the potential underlying mechanisms that have been proposed. A greater understanding of the relationship between HRV infections, allergic inflammation, and asthma could lead to new strategies to prevent and treat childhood asthma.
HRV and host innate immune responses
HRVs, members of the family Picornaviridae, are positive-sense, single-stranded RNA viruses with a genome of approximately 7.2 kb. Until recently, HRVs have been classified into 2 species, HRV-A (containing 74 serotypes) and HRV-B (containing 25 serotypes), based on phylogenetic sequence criteria10). The development of highly sensitive molecular techniques to detect HRV in clinical specimens led to the identification and designation of a novel species, HRV-C. More than 60 types of this new species have since been recognized11).
HRVs are also classified on the basis of receptor specificity. More than 90% of HRV serotypes, known as the “major group,” utilize the intercellular adhesion molecule 1 (ICAM-1) as a receptor to enter host cells1213), whereas the “minor group” of HRVs utilizes the low-density lipoprotein receptor (LDLR). The major group consists of both HRV-A and HRV-B species, whereas all members of the minor group are HRV-A viruses based on phylogenetic classification. The receptor for HRV-C viruses is currently unknown. HRV enters host cells via endocytosis mediated by these receptors, and the process can be clathrin-dependent or independent, depending on the receptor type14). Once HRV enters host cells, pattern recognition receptors (PRRs) within the airway recognize HRV, which induces proinflammatory and antiviral responses. These PRRs include Toll-like receptors (TLRs), retinoic acid inducible protein I (RIG-I), and melanoma differentiation associated gene 5 (MDA5). In the endosome, TLR3 and TLR7/8 recognize dsRNA intermediates generated during the HRV replication cycle and ssRNA, respectively. TLR7/8 may be induced upon HRV infection, but currently TLR3 seems to be the dominant PRR for HRV. Detection of dsRNA intermediates by TLR3 triggers upregulation of cytoplasmic RIG-I and MDA5, which also recognize newly synthesized ssRNA and dsRNA intermediates in the cytoplasm1516) (Fig. 1). Subsequently, these 2 PRRs stimulate the interferon (IFN) gene and the expression of proinflammatory cytokines, such as RANTES, IFN-γ-induced protein 10 (IP-10), and IL-81517). These cytokines modulate proliferation, activation, and recruitment of inflammatory cells to the airway. Specifically, IL-8, a chemoattractant and activator of neutrophils, is known to be a major determinant of clinical outcomes of HRV infection. Bronchial epithelial cells (BECs) produce type I IFN-β and type III IFN-λs upon HRV infection151618). These 2 IFN families exhibit very similar properties. Both IFN-β and IFN-λ signals through their respective receptors initiate transcription of IFN-stimulated genes (ISGs)1920). The ISGs modulate HRV elimination by degrading viral RNA, preventing virus-associated protein trafficking and virion assembly, and inducing apoptosis. Limited studies suggest that a key difference between these 2 IFN families could be the cell specificity of the responses to each. Unlike the type I IFN response, which occurs in most cell types, the response to IFN-λ appears to be restricted to epithelial cells, suggesting that IFN-λ plays a dominant role in protection against viral invasion through skin and mucosal surfaces21).
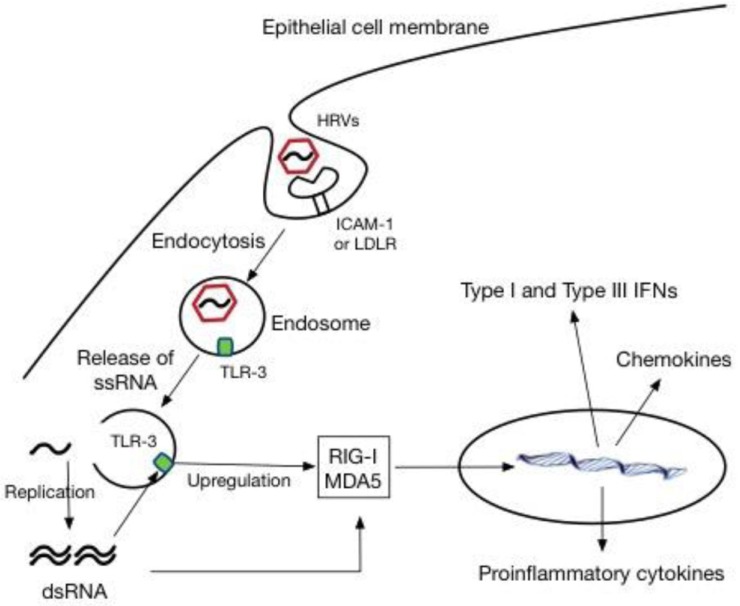
Human rhinovirus (HRV) infection and innate immune responses in epithelial cells. ICAM-1, intercellular adhesion molecule 1; LDLR, low-density lipoprotein receptor; TLR-3, Toll-like receptor-3; ssRNA, single-stranded RNA; dsRNA, double-stranded RNA; RIG-I, retinoic acid inducible protein I; MDA5, melanoma differentiation associated gene 5; IFN, interferon.
Murine model of HRV infection
Until recently, the lack of small animal models of HRV infection has been a major obstacle to the investigation of the pathogenesis of HRV infection. One of the main issues confronting mouse model development was that HRVs have a high degree of species specificity. To our knowledge, there are no known murine rhinoviruses, and the major group HRVs cannot bind to ICAM-1 from other species, with the probable exception of chimpanzees and gibbons2223). However, transfection of chimeric human/mouse ICAM-1 in vitro rendered mouse respiratory cells susceptible to HRV16, a major group HRV, infection. In contrast to major group viruses, minor group viruses can attach to mouse LDLR, but only HRV-1A and HRV-1B can replicate in mouse cells24). Based on the results from these in vitro studies, practical mouse models of major and minor group HRV infection were introduced in 200825). One limitation to these murine models is that HRV titers decline rapidly within 24 hours after inoculation, preventing investigations of viral replication and airway dysfunction beyond this time point. Nonetheless, the development of rodent models of HRV infection has provided a useful tool to examine the immunopathogenesis of HRV and to understand the role of HRV in the development and acute exacerbation of asthma in vivo.
Role of HRV infection in asthma development
Although asthma can arise at any age and its phenotypic expression can vary over time, asthma often originates early in life26). In addition to individual host factors, environmental exposures have a major impact on the development of asthma. There is little doubt that respiratory viral infections constitute an important environmental risk factor1320). The most common viruses associated with wheezing illnesses early in life are RSV and HRV27). Multiple epidemiological studies have demonstrated that severe RSV bronchiolitis is associated with subsequent, persistent wheezing and childhood asthma456). With advancements in molecular assays to detect respiratory viruses, it has become apparent that HRV infections during infancy are a strong predictor for the development of asthma28).
In this regard, the Childhood Origin of Asthma (COAST) study has provided valuable insight into the association between early life HRV infection and subsequent asthma development29). COAST is a high-risk birth cohort study investigating the role of immune response aberrations and respiratory viruses in the development of asthma and other allergic diseases. Data from this cohort showed that at least one wheezing illness with rhinovirus infection during infancy was the most significant risk factor for persistent wheezing at the age of 3 years30). Furthermore, HRV wheezing illness in the first 3 years of life was a much stronger risk factor for the development of asthma at the age of 6 years than RSV wheezing illness or aeroallergen sensitization6). Another high-risk birth cohort study in Australia reported similar findings: HRV wheezing illness during the first year of life was associated with increased asthma risk at the age of 5 years, although this association was restricted to children with aeroallergen sensitization by the age of 2 years31). However, it should be noted that these studies are based on outpatient cohorts of infants that are at high risk for developing asthma; therefore, these findings may not be applicable to the general population.
There are limited data available on the association between hospitalization with severe HRV wheezing illness and persistent wheezing or future asthma in the general population. Recent studies in Europe provide the information about this question. An Italian cohort study demonstrated that the rate of recurrent wheezing was higher after hospitalization with HRV bronchiolitis than with other respiratory viruses during a 1-year follow-up period32). HRV infection and a positive family history for asthma were independent risk factors for recurrent wheezing in this cohort. A case control study in Finland also demonstrated that infants hospitalized with HRV wheezing had the highest risk for subsequent asthma, and this relationship persisted at least through the adolescence33). Taken together, these studies suggest the importance of early life HRV infection as a major risk factor for recurrent wheezing illness and future asthma inception.
Although virtually all children (>90%) are infected with HRV during infancy34), the factors that predispose a subset to develop persistent wheezing illnesses or asthma are not fully understood. Individual host factors, such as genetic factors and atopy, most likely interact with HRV infections in susceptible patients, resulting in an increased risk for asthma development. Evidence regarding genetic susceptibility in HRV infection is beginning to emerge. Genome-wide association studies (GWAS) have consistently linked the asthma susceptibility locus 17q21 to nonatopic childhood-onset asthma3536). Data from 2 birth cohort studies, COAST and Copenhagen Studies on Asthma in Childhood, demonstrated that 17q21 variants were associated with HRV wheezing illness in early life, but not with RSV wheezing illnesses. Children with the risk-associated genetic variants only developed asthma after wheezing with HRV infection9). Although the disease-associated variants at this locus impact the expression levels of 2 17q21 genes, GSDMB and ORMDL3, the functions of the products encoded by these genes in the pathogenesis of asthma have not been well understood. Recently, however, potential roles of ORMDL3 in asthma pathogenesis have been identified. These included dysregulation of the unfolded protein response, leading to airway inflammation and remodeling through sarco/endoplasmic reticulum Ca2+ ATPase, and negative regulation of sphingolipid synthesis resulting in airway hyperresponsiveness373839). In contrast to the results from the birth cohort studies, recent in vitro studies have suggested that ORMDL3 might have an antiviral effect, rather than increase susceptibility to HRV infection. Overexpression of ORMDL3 in human airway epithelial cells increased the expression of 2′-5′ oligoadenylate synthase38), a molecule that inhibits viral replication by degrading viral RNA through RNase L activation40) (Fig. 2). Moreover, the expression level of ORMDL3 was significantly increased in HRV-stimulated peripheral blood mononuclear cells9). Therefore, the exact role of ORMDL3 in HRV infection needs to be clarified by further epidemiological and experimental studies.
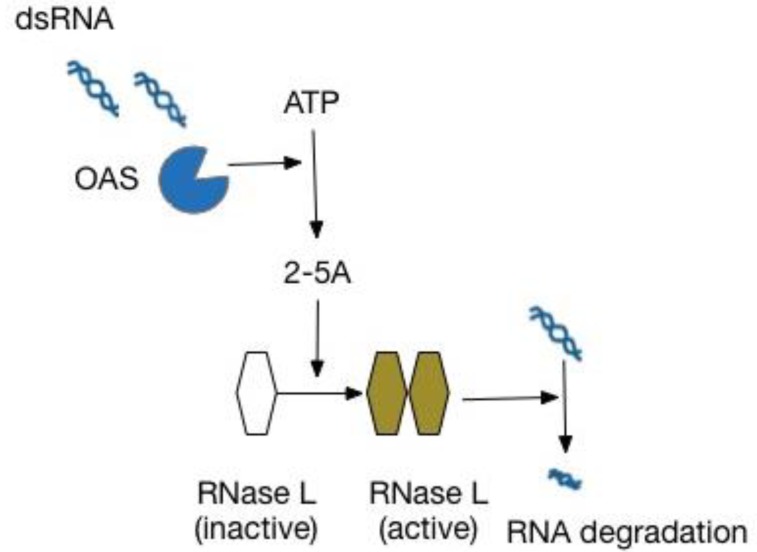
The OAS/RNase L pathway in an innate immune response against viruses. dsRNA, double-stranded RNA; OAS, 2′-5′-oligoadenylate synthetase; 2-5A, 5′-phosphorylated, 2′-5′-linked oligoadenylates; RNase L, 2′-5′-oligoadenylate-dependent ribonuclease L.
Regarding genetic susceptibility to asthma and HRV infection, a recent GWAS identified CDHR3 (encoding cadherin-related family member 3), which is highly expressed in the airway epithelium, as a new susceptibility locus for early childhood asthma41). A subsequent in vitro study suggested that mutations in this susceptibility gene could be a risk factor for increased HRV wheezing illness by mediating HRV-C entry into host cells42). HRV-C has been identified as the main pathogenic HRV species associated with infant wheeze, hospitalization, and subsequent development of asthma in recent research43). Together, this evidence suggests that HRV infection is associated with asthma susceptibility in genetically predisposed children.
Another potential host factor involved in promoting asthma inception via interaction with HRV infection is atopy. Early sensitization to allergens has consistently been identified as a major risk factor for asthma development. However, the temporal sequence of these 2 events has not been clear. Recently, however, several studies have suggested that allergic sensitization may precede HRV wheezing illness, while HRV wheezing does not increase the risk of subsequent allergic sensitization444546). A number of mechanisms by which allergic sensitization might increase the risk of HRV-induced wheezing have been proposed. Th2 cytokines, such as interleukin (IL)-4, IL-5, and IL-13, further upregulate the expression of ICAM-1, the receptor for 90% of HRV serotypes, on the surface of epithelial cells in the respiratory tract47), while HRV infection itself increases ICAM-1 expression48). This response may render epithelial cells more susceptible to HRV infection. Moreover, the epithelial inflammatory response to HRV is downregulated in an atopic environment, resulting in increased viral replication and cell damage, whereas transforming growth factor is upregulated49). These results suggest one possible mechanism by which HRV infections could promote airway remodeling. Allergic inflammation may directly inhibit host antiviral responses. An ex vivo model demonstrated that plasmacytoid dendritic cells (pDCs), key antigen-presenting cells recruited to the airway during viral infections, from patients with asthma secreted significantly less IFN-α upon exposure to influenza, and IgE cross-linking prior to viral challenge resulted in the abrogation of a virus-induced pDC IFN-α response50). This counterregulation between IgE-mediated pathways and antiviral responses might increase the severity of disease following viral infection in allergic individuals.
Supporting the “2-hit hypothesis” of asthma development, these observations have led to the notion that genetic predisposition combined with HRV infection may promote asthma in children51). Further research is needed to explore host factors and additional environmental factors that modify the risk for asthma following early life HRV infection and the mechanisms that underlie the interactions between HRV infection and these factors.
Impacts of HRV infection on asthma exacerbation
Respiratory viral infection has been recognized as the most common cause of asthma exacerbation5253). A clear temporal relationship between outbreaks of upper respiratory viral infection and increased hospitalizations for asthma exacerbations is consistently observed in the northern hemisphere in the spring and also early fall, with a peak in September5455). This seasonal pattern is sometimes referred to as the “ September epidemic.” Epidemic peaks of asthma exacerbations in adults are also observed shortly after children return to school and at the peak time of year for HRV infections, suggesting viral infections as a cause.
Before the advent of polymerase chain reaction (PCR) assays, the role of viral infections in asthma exacerbation was underappreciated. Molecular assays to detect respiratory viruses changed our understanding of asthma exacerbations. PCR-based studies investigating the prevalence of viral infection during asthma exacerbations showed that upper respiratory viral infections were associated with up to 85% of exacerbated asthma cases in children, and approximately 50% of these cases in adults54565758). Among viruses detected in these studies, HRV was the dominant pathogen, making up about 60% of the viruses detected5960). Moreover, HRV was also the most common viral pathogen detected in children and adults hospitalized for asthma exacerbations. While the association is clear, the molecular and cellular events behind HRV-induced asthma exacerbations remain unclear.
A number of mechanisms by which HRV might exacerbate asthma have been proposed. Once, it was assumed that HRV could not infect the lower airway, because the optimal temperature for HRV replication was determined to be 33℃–35℃ in early studies6162). Then the common explanation was that inspiration of dry and cold air through mouth due to nasal blockage triggered asthma symptoms. Nasal-bronchial reflex and release of proinflammatory mediators from the nose into the circulation have been also proposed as underlying mechanisms.
However, it is now apparent, from subsequent in vitro and in vivo studies, that HRV can infect the lower airways. HRV has been detected in the lower airways via immunohistochemistry, reverse transcription-, and in situ hybridization of positive-strand viral RNA636465). Interestingly, in contrast to influenza and other respiratory viruses, HRV infects relatively few epithelial cells in the airway and causes minimal cytotoxicity6667). Moreover, the amount of virus shed and the extent of epithelial damage does not correlate with the severity of symptoms68). Together, these studies suggest that HRV provokes symptoms through virus-specific cytopathic effects, rather than through direct cytotoxic effects.
One possible explanation for these findings is that HRV infection induces proinflammatory responses in airway epithelial cells, the primary site of HRV infection and replication. Studies using cultured human airway epithelial cells demonstrated that HRV infection induced the expression of a wide range of cytokines (IL-1β, IL-6, IL-11), growth factors (G-CSF, GM-CSF), and chemokines (CXCL8, CXCL5, CXCL10, RANTES) that can contribute to the activation and recruitment of inflammatory cells to the airway6970). These cytokines were also increased in airway secretions during in vivo HRV infections71). These molecules can be expected to enhance airway inflammation and potentiate preexisting allergic inflammation.
In addition to the pro-inflammatory cytokines mentioned above, HRV infection can trigger the release of Th2-promoting cytokines, such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), from airway epithelial cells. These cytokines have been recognized as critical elements in type 2 T-cell differentiation. Recently, a number of studies have provided information about the role of these epithelium-derived cytokines in HRV-induced asthma exacerbation. Human in vitro and in vivo HRV infection studies have demonstrated a heightened intrinsic capacity for IL-25 expression in HRV-infected asthmatic BECs and increased IL-25 level in the nasal fluid of atopic asthmatics in experimental HRV infections. Similar findings were observed in a murine model of HRV-induced asthma exacerbations. In addition, blocking the IL-25 receptor reduced type 2 cytokine expression, mucus production, and recruitment of inflammatory cells72). Another human experimental HRV infection model revealed increased levels of IL-33 and type 2 cytokines in the bronchial mucosal lining fluid of asthmatics during experimental HRV infection, and the levels of these cytokines were related to the severity of exacerbation. The production of these cytokines by peripheral blood T cells and type 2 innate lymphoid cells from the same subjects was induced when cultured with the supernatants of HRV-infected BECs and this induction was entirely dependent upon IL-3373). TSLP is also considered a potential molecular link between HRV infection and the characteristic Th2 response seen in asthma exacerbation. An ex vivo model using asthmatic BECs revealed that stimulation with dsRNA, a surrogate of viral infection, induced exaggerated TSLP production, accompanied by reduced IFN-β production74). Furthermore, lung tissue TSLP was induced by both HRV infection and exposure to dsRNA in a murine model of allergic asthma75). Taken together, these observations suggest a potential role of epithelial mediators in HRV-induced asthma exacerbation.
IFNs play a critical role in the host defense against respiratory viruses, including HRV. Type I (IFN-α/β) and type III (IFN-λ) INFs are the most significant IFNs secreted by the airway epithelium during acute HRV infection2176). It has been suggested that impaired epithelial production of IFN-β and IFN-λ in asthmatic subjects may contribute to viral exacerbation of asthma7778). Recent studies investigating airway interferon production in response to HRV in children have demonstrated reduced IFN secretion from BECs after HRV stimulation in asthmatic and atopic nonasthmatic children compared with IFN secretion seen in nonatopic, nonasthmatic children79). Additionally, ex vivo IFN-λ and IFN-β induction correlated inversely with severity of symptoms, bronchoalveolar lavage viral load, and airway inflammation, and correlated positively with reductions in lung function during in vivo HRV infection78). However, the association between IFN response and HRV infection in asthmatic children seems to be more complex. A prospective study in school-aged children with asthma showed increased IFN-λ1 production in wheezing children with HRV infection as compared to nonwheezing children with HRV infection, suggesting that a dysregulated IFN response rather than a simple deficiency of IFN production mediates HRV-induced asthma exacerbations80). Further research is needed to define role of IFN-λ in HRV-induced asthma exacerbation and the mechanism of this IFN dysregulation.
HRV infections might also contribute to asthma exacerbations via enhanced mucus production. In vitro studies have shown that HRV infection of human airway epithelial cells increased mRNA expression of mucin-2 (Muc2), mucin-3 (Muc3), mucin-5 subtypes A and C (Muc5AC), mucin-5 subtype B (Muc5B), and mucin-6 (Muc6). MUC5AC and total mucin concentrations increased in supernatants and lysates of the surface cells 81). Supporting this in vitro study, a murine model of HRV infection demonstrated that RV1B induced Muc5AC and Muc5B mRNA in the lung, and increased MUC5AC and MUC5B protein in BAL25).
HRV infections can affect the contractility of airway smooth muscle cells, which might contribute to asthma exacerbations. Limited in vitro and animal studies have revealed that binding of HRV to its receptor, ICAM-1, on airway smooth muscle cell surfaces can enhance the contractility of airway smooth muscle cells and impair the response to β-adrenergic agonists without any cytopathic effects or viral replication8283). Moreover, several in vitro studies have demonstrated that HRV can infect a variety of other cell types, including fibroblasts, monocytes, and macrophages, although the contributions of these individual cell types to asthma exacerbation are still not well understood. Further research is needed to define the role of these cells in HRV-induced asthma exacerbation.
Conclusions
The advent of molecular diagnostics to detect respiratory viruses has led to new insights into the role of HRV infections in the development and acute exacerbation of asthma in children. Recent clinical and experimental studies have identified plausible pathways by which HRV infection could be causal, particularly in a susceptible host and trigger disease exacerbation. Airway epithelial cells, the primary site of infection and replication of HRV, play a key role in these processes. Additional in vitro and in vivo studies using murine model of HRV infection with genetically modified mouse may help to improve our understanding of the interplay between genetic factors and HRV infection in the development of childhood asthma and to identify novel targets for therapeutic intervention.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.