


Introduction
Macrophage activation syndrome (MAS) is characterized by fever, hepatosplenomegaly, severe cytopenia, serious liver disease, and coagulopathy1). MAS is a severe, potentially fatal condition associated with excessive activation of immune cells, leading to hypercytokinemia and overwhelming systemic inflammatory reactions with hemophagocytosis due to defective immune regulation1,2). MAS is used to describe secondary hemophagocytic lymphohistiocytosis (HLH) associated with autoimmune disease. Primary HLH developed in genetic disorder, but secondary HLH is triggered by various etiologies including infection, medication, malignancies, and autoimmune diseases1,2,3).
In autoimmune disease, hypercytokinemia in systemic inflammatory response may induce MAS. The annual incidence of HLH in children was reported as 1 in 800,0004). About 10% of secondary HLH, called as MAS, were associated with autoimmune disorders4), and MAS occurred frequently in 10%ŌĆō20% of systemic juvenile idiopathic arthritis4,5). Recurrent MAS or HLH was developed in underlying genetic disorders, but is rare in secondary cause2,6). MAS in ankylosing spondylitis (AS) was very rare in literature review5,7).
We reported a case of recurrent MAS in AS since toddler age, although we could not identify etiology of recurrent HLH until adolescent age.
Case report
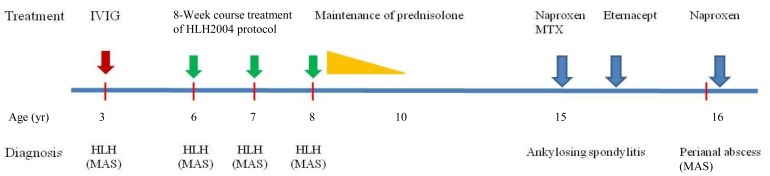
A 16-year-old boy was transferred from Department of Surgery due to remittent fever with pancytopenia and splenomegaly despite of improvement of septic shock without any organisms. He had received fistulectomy with colostomy due to intractable perianal abscess 2 months previously. He had been diagnosed with HLH according to HLH 1994 guideline at 3 years of age8), and had been treated with intravenous immunoglobulin (IVIG). HLH recurred 3 years later, and he was treated according to HLH 2004 protocol including dexamethasone, etoposide, and cyclosproin A9), and remained symptoms free without maintenance therapy. He relapsed again at ages 7 and 8, but we were unable to identify any causes. He received maintenance prednisolone treatment for 2 years after 4th attack.
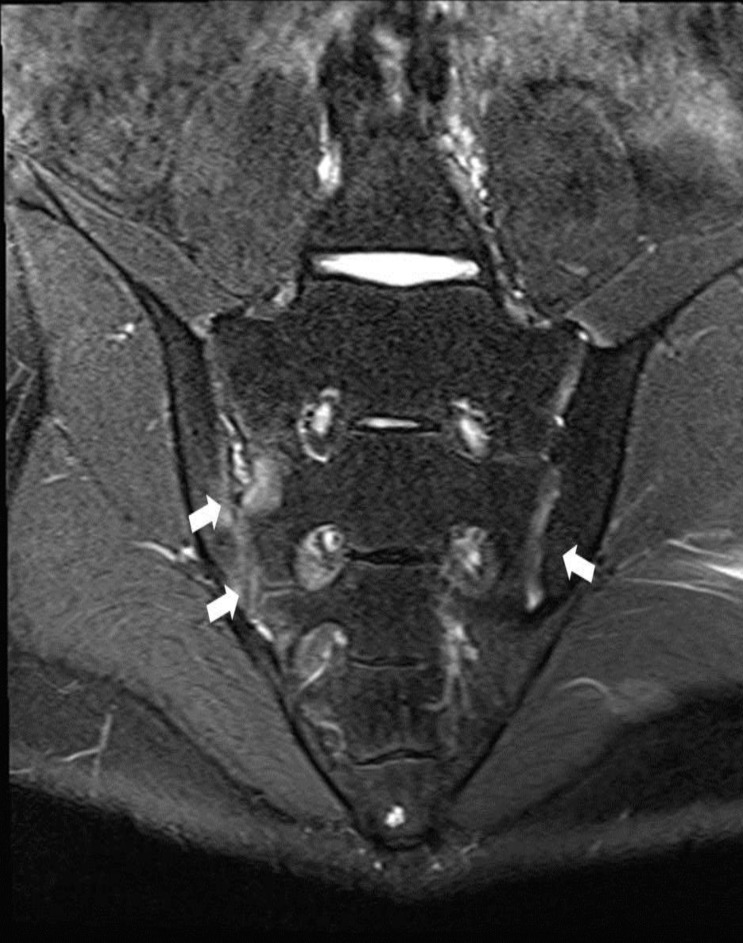
He was symptom free until 15 years of age. He complained back and both knee joints pain with right ankle swelling for 1 month at June, 2012. We performed ultrasonography for synovial fluid from joints. Sonographic findings showed profound amount of fluid collection on suprapatellar recess and synovial hypertrophy. We aspirated about 50 mL of synovial fluid with 10,000/┬ĄL of white blood cell (WBC, 66% segmented neutrophil, 16% lymphocyte, 18% monocyte). Human leukocyte antigen (HLA) B27 was positive in genetic study of patient. Magnetic resonance imaging findings for sacroiliac joints (SI) showed both SI joint cartilage abnormalities with erosion of right SI and bone marrow edema. He was diagnosed with HLA B27 positive juvenile AS compatible with bilateral sacro-iliitis (Fig. 1). We began medication with naproxen and methotrexate, but showed symptom aggravation. His symptoms remained with 4.75 of Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) at 4 month later. He did not complaint pain after Eternacept treatment. His BASDAI was 1.6 before development of intractable perianal abscess. He was admitted to department of surgery for surgical intervention with fistulectomy and colostomy. However, he developed fever and signs of septic shock, and was treated with antibiotics and IVIG.
On physical examination, he was febrile with splenomegaly. His wound was clean state without any inflammation finding. His laboratory data showed WBC count 1,240/┬ĄL, platelet 44,000/┬ĄL, ferritin 2,707 ng/mL, triglyceride 343 mg/dL, aspatate transaminase 238 IU/L, alanine transaminase 145 IU/L, and fibrinogen 96 mg/dL. We performed bone marrow aspiration and biopsy, which showed histiocytic hyperplasia with hemophagocytosis. There was no serologic evidence of any viral infections including Epstein-Barr (EB) virus. We diagnosed with MAS from HLA B27 positive AS. He received naproxen only, and improved without any other immunomodulatory medication. Fever was dropped after medication, and his symptoms were improved, even though persistent splenomegaly. Also, his laboratory data returned to near normal range within 4 weeks. We evaluated gene study for primary HLH and inflammasome, and got no any genetic abnormalities2,6). We described his clinical course and treatments of MAS and JAS since 3 years old as Fig. 2.
This study was approved by Institute of Review Board from Seoul St. Mary's Hospital, College of Medicine, The Catholic University of Korea (KC16ZISE0297).
Discussion
The clinical criteria for HLH include many nonspecific findings that overlap with severe inflammatory conditions1,2). Hemophagocytosis is a pathologic finding that activated macrophages phagocyte blood cells due to inappropriate immune response1,2,4). The causes of HLH are due to genetic defects (primary) or various etiologies including infections or autoimmune diseases (secondary)3,8,9). MAS is called as acute systemic inflammation from autoimmune disease or autoinflammatory conditions including systemic juvenile idiopathic arthritis or systemic lupus erythematosus3,5,10). MAS is rarely reported in AS5,7). In our case, an adolescent boy experienced recurrent HLH controlled by immunosuppressive agents since toddler age. He was diagnosed with JAS at 15 year-old.
The diagnostic criteria of HLH was based on HLH-2004 diagnostic guideline, as follow as presence of a molecular diagnosis consistent with HLH including Munc gene or meeting 5 of 8 clinical and laboratory diagnostic criteria including fever, hepatosplenomegaly, cytopenia, hypertriglyceridemia/hypofibrinogenemia, hemophagocytosis, low or absent Natural Killer (NK) cell activity, ferritin over 500 ng/mL, and soluble CD25 over 2,4009). The preliminary diagnostic guidelines of MAS in autoimmune disease consisted of at least 2 laboratory criteria or the presence of at least one laboratory criterion including cytopenia, elevated levels of AST and lactic dehydrogenase, hypofibrinogenemia, hyperferritinemia, hypertriglyceridemia, and one clinical criterion including fever, hepatosplenomegaly, hemorrhagic manifestations, and central nervous system dysfunction3). He was diagnosed with MAS after JAS, compatible findings with fever, cytopenia, splenomegaly, hyperferritinemia, hypertriglyceridemia, elevated with AST, and hypofibrinogenemia.
Juvenile ankylosing spondylitis (JAS) is included in the enthesitis related arthritis, and diagnosed with over 6-week symptoms and 8 years by International League of Association for Rheumatology11). JAS is a strong genetic predisposition as the high frequency of positive HLA B27. The prevalence of AS is around 0.2% in general population12), and symptoms developed only 10%ŌĆō20% before age 1611). The pathogenesis of JAS was related with HLA B27, and autoreactive CD8 T cells recognized peptide presentation on HLA B27 heavy chain misfolding, and produced various cytokines including TNF-╬▒ and IL-113,14). These cytokines induced inflammation to joints and new bone formation14). He complained back and joint pain over 6 weeks at 15 years old, and was diagnosed with JAS according to sacro-iliac inflammation and positive HLA B27. We did not evaluated HLA B27 until joint symptoms developed.
This patient experienced recurrent HLH since toddler age, but he was tolerable with low dose steroid after HLH-94 or HLH 2004 treatment protocol8,9). The result of negative perforin gene suggested that his clinical course was fortunately not fatal. When he relapsed HLH, we evaluated etiology including viral infections, and did not identify. The patient with relapsed HLH received chemotherapy or hematopoietic stem cell transplantation according to HLH treatment protocol8,9). However, he was very tolerable in spite of low dose steroid. In diagnostic criteria of JAS, joint symptoms were developed since at least 8 years old11). He was diagnosed with JAS at 15 years old. In this patient, recurrent HLH might be related with underlying JAS pathogenesis. In our case, HLA B27 would be contributed to immune dysregulation for CD8 T cell activation13,14,15). The autoreactive CD8 T cells might produce profound cytokines and induce clinical manifestations of HLH1,2,5). This immunologic background might be related with recurrent HLH since toddler age.
For identifying underlying genetic autoinflammatory disease, we evaluated genetic study for inflammasome because of recurrent MAS6). We did not find out any genetic abnormalities related with NLRC4. Therefore, immunologic aspect with recurrent MAS might be only associated with HLA B27 in this patient, respectively.
We reported that an adolescent boy with JAS experienced recurrent MAS since toddler age. We suggest that underlying autoimmune disease should be considered as the cause of recurrent MAS in a young patient after primary HLH has been excluded.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


