Alagille syndrome and a JAG1 mutation: 41 cases of experience at a single center
Article information
Abstract
Purpose
Alagille syndrome is a complex hereditary disorder that is associated with cardiac, hepatic, skeletal, ocular, and facial abnormalities. Mutations in the Notch signaling pathway, such as in JAG1 and NOTCH2, play a key role in embryonic development. A cardiac or hepatic presentation is a critical factor for determining the prognosis.
Methods
We conducted a retrospective study of 41 patients with Alagille syndrome or a JAG1 mutation between 1983 and 2013.
Results
The first presentations were jaundice, murmur, cyanosis, and small bowel obstruction at a median age of 1.0 months (range, 0-24 months). The JAG1 mutation was found in 27 of the 28 genetically-tested patients. Cardiovascular anomalies were identified in 36 patients, chronic cholestasis was identified in 34, and liver transplantation was performed in 9. There was no significant correlation between the severity of the liver and cardiac diseases. The most common cardiovascular anomaly was peripheral pulmonary stenosis (83.3%), with 13 patients having significant hemodynamic derangement and 12 undergoing surgical repair. A total bilirubin level of >15 mg/dL with a complex surgical procedure increased the surgical mortality (P=0.022). Eight patients died after a median period of 2.67 years (range, 0.33-15 years). The groups with fetal presentation and with combined severe liver and heart disease had the poorest survival (P<0.001).
Conclusion
The group with combined severe liver and heart disease had the poorest survival, and a multidisciplinary approach is necessary to improve the outcome.
Introduction
Alagille syndrome (AGS) was first reported by Alagille et al.1) in 1969. The main clinical presentations are chronic cholestasis characterized by the paucity of intrahepatic bile ducts, congenital cardiac anomaly involving pulmonary stenosis, butterfly-like vertebral abnormalities, ocular changes such as posterior embryotoxon, and peculiar facial abnormalities (broad forehead, deep-set and widely spaced eyes, small pointed chin, and saddle or straight nose)234). The clinical criteria for diagnosis meet 3 out of 5 main clinical presentations. There have been several reports evaluating the outcome of liver disease in patients with AGS5). To date, there have been limited published studies investigating the characteristic cardiac features of AGS and the outcome of congenital cardiac defects with AGS in Korea.
AGS confirmed the pathognomonic gene mutation of the Notch signal pathway; more than 97% of patients had mutation of the JAG1 gene, and <1% had the mutation at the NOTCH2 gene67). The Notch signal pathway has a main role in organogenesis, especially the cardiac valve, cardiac outlet tract, and ventricular septum8). The inheritance is autosomal dominant9). In response to the increasing significance of liver and heart presentations in patients with AGS, the necessity of cardiac evaluation has been questioned.
We conducted this study to determine: (1) the clinical characteristics of cardiovascular abnormalities in AGS; and (2) the correlation between cardiac and hepatic manifestation and the factors affecting the prognosis of AGS patients.
Materials and methods
1. Patients
We investigated 49 patients who visited Seoul National University Hospital for evaluation of murmur/cyanosis or cholestasis, between 1983 and 2013. We excluded 8 cases; 7 suspected AGS patients who were lost to follow-up until diagnosis, and 1 suspected AGS patient who died before the diagnosis was confirmed. Finally, 41 patients who were diagnosed with AGS were included in the study. The inclusion criteria were as follows: (1) positive for ≥3 clinical criteria (chronic cholestasis, congenital heart disease, butterfly vertebrae, posterior embryotoxon, and peculiar face); and (2) positive for ≥1 clinical criteria with a proven JAG1 mutation3710).
The medical records that were reviewed retrospectively included the ophthalmologists' physical findings and radiologists' interpretations of plain radiographs showing the vertebrae. We investigated the gene mutations of the JAG1 gene and reviewed the echocardiography/ cardiac catheterization records.
Chronic cholestasis was defined as a prolonged serum direct bilirubin level of ≥1.0 mg/dL for ≥6 months. We obtained the liver biopsy findings via ultrasonography-guided biopsy or wedge resection. To evaluate the existence of hepatocellular carcinoma, we investigated the radiologic readings and the serum alpha-fetoprotein in patients with suspected AGS. Severe liver disease was defined based on the necessity of invasive intervention that was indicated by one or more of the following: (1) serum total bilirubin level >15 mg/dL; (2) who had liver cirrhosis/liver tumor; (3) who underwent liver transplantation; and (4) Kasai operation. The cutoff value of serum total bilirubin refers from the mean of our study group (mean, 14.8±16.0 mg/dL; median, 7.15 mg/dL; range, 0.5-70.4 mg/dL).
Cardiovascular presentations were classified into primary and combined anomalies. Furthermore, cardiac anomalies were categorized into right/left, and "other" side-the lesion that was neither right-sided nor left-sided. Severe heart disease was defined in cases of the following: (1) right-sided lesions such as tetralogy of Fallot, double outlet of the right ventricle, severe pulmonary stenosis with a pressure gradient of >40 mmHg, or a right/left ventricle (or aorta) pressure ratio of >0.6 in catheterization; (2) left-sided lesions such as coarctation of the aorta or interruption of the aorta or severe aortic stenosis with a pressure gradient of >40 mmHg; and (3) hypertrophic cardiomyopathy. The severe heart disease was defined based on the need for intervention or the critical condition having heart failure, hypoxemia, or hemodynamic instability. The complexity of the cardiac operation was determined using the Aristotle score11).
2. Statistical analysis
Data are presented with mean±standard deviation, median (range). Categorical data were compared using the chi-square test with Yate's correction if indicated. Continuous data were compared using the t test. Survivorship analysis was performed according to the Kaplan-Meier method, with right-censored data for no event or missing data. A P value of <0.05 was considered significant. All calculations were performed using IBM SPSS Statistics ver. 19.0 (IBM Co., Armonk, NY, USA)
The study was approved by the Institutional Review Board of Seoul National University Hospital (IRB No. H-1405-098-580).
Results
1. Demographic data
The study population comprised 23 boys and 18 girls. Of the 41 patients enrolled, 35 met ≥3 clinical criteria, and 6 met ≥1 clinical criteria with a proven JAG1 gene mutation. With regard to the clinical criteria, 34 patients (82.9%) displayed chronic cholestasis, 36 (87.8%) displayed congenital heart disease, 37 (92.5%; except for one, which was undetermined) displayed a peculiar face, 13 (31.7%) displayed butterfly vertebrae, and 12 (30.0%; except for one, which was undetermined) posterior embryotoxon. An analysis of the JAG1 gene was performed in 28 patients, with 27 subjects showing the JAG1 gene mutation, and one subject showing several polymorphisms but no mutations.
The patients had manifested first features as follows: jaundice (58.5%), murmur (7.3%), cyanosis (4.9%), jaundice with murmur (12.2%), jaundice with small bowel obstruction (2.4%), and cardiac anomaly detected during the prenatal period via fetal echocardiography (14.6%). The median age of onset was 1.0 months (range, 0-24 months), with 6 fetal presentations (14.6%), 22 at 0-30 days (53.7%), 11 at 1-4 months (26.8%), 1 at 5-12 months (2.4%), and 1 at >12 months (2.4%). There was no significant difference in age of onset between the patients who had presenting jaundice and those who had presenting cardiac features such as fetal cardiac anomaly or cyanosis or cardiac murmur (2.03±4.43 months vs. 0.91±2.03 months, respectively, P=0.34).
2. Liver evaluation
Overall, 37 of the 41patients underwent liver biopsy via ultrasonography guidance or wedge resection. Thirty-two patients (78 %) had findings compatible with a paucity of intrahepatic bile ducts. Moreover, one of them had documented biliary atresia. The median level of maximum total bilirubin was 7.15 mg/dL (range, 0.5-70.4 mg/dL). The severe liver disease group consisted of 19 patients (46.3%), and 9 of them (47.7%) received liver transplantation at the median ages of 1.92 years (range, 1.25-23.08 years). The patient who underwent living donor liver transplantation at first underwent a second transplantation of a cadaveric donor liver after 8 months because of biliary cast syndrome following liver transplantation; however, the patient died on the day of surgery because of hyperacute rejection leading to liver failure. During the follow-up period of 10 years, 12 patients survived; the 5-year survival rate after liver transplantation was 33.3%.
3. Cardiac presentation
According to the echocardiography or catheterization, 36 patients (87.8%) had cardiac anomalies (Table 1). The right sided lesions were found in 30 and the left side lesions in 5 patients. The most common cardiac anomaly was pulmonary stenosis (97.2%, 35 of 36), with the involved lesion type being, peripheral pulmonary tree stenosis in 25 patients (71.4%), valvular pulmonary stenosis in 5 patients (14.3%), supravalvular pulmonary stenosis with peripheral stenosis in 3 patients (8.6%), and valvular pulmonary stenosis with peripheral stenosis in 2 patients (5.7%). Thirteen patients were assessed for severe heart disease which required surgical or transcatheter intervention. There was no statistically significant correlation between the severity of liver disease and the severity of cardiac disease (P=0.511).

Cardiovascular anomalies
4. Cardiac intervention
Of the 36 patients, 12 patients underwent cardiac catheterizations, 6 underwent diagnostic procedures, 4 underwent balloon angioplasty, and 2 underwent percutaneous valvuloplasty procedures. There were a mean of 1.83 catheterizations per patient, which were performed at a median of 3.25 years (range, 0.83-21.67 years). The major of diagnostic catheterization cases (83%) had proceeded as preoperative evaluations. The balloon angioplasty procedure was performed 1-5 times in each patient. The cutting balloon angioplasty procedure was performed 4-5 times for diffuse pulmonary branch stenosis; however, it did not result in significant regression of stenosis. Those cases received an additional balloon angioplasty procedure despite of the angioplasty operation. One patient was tetralogy of Fallot with pulmonary atresia. He confirmed the JAG1 mutation. He underwent total 4 times of balloon angioplasty. Initial balloon angioplasty procedure was for stenosis after pulmonary angioplasty operation. After that, several procedures followed without reduction of the apparent pressure gradient. Another patient was diagnosed as peripheral pulmonary stenosis which was categorized to severe heart disease for right/left ventricle (or aorta) pressure ratio of >0.6. He confirmed the JAG1 mutation. For regression of severe peripheral pulmonary stenosis, he underwent total 5 times of balloon angioplasty including cutting balloon procedures. The first procedure was done at 3 years of age. The 6th procedure was planned because of right ventricular hypertrophy and restenosis of pulmonary artery.
Cardiac surgery was performed in 12 patients. The patients underwent a mean of 1.83 surgeries (range, 1-5 surgeries) at a median age of 1.25 years (range, 0.02-8.83 years). The patient who underwent 5 surgeries was diagnosed with tetralogy of Fallot with pulmonary atresia. He was mentioned earlier as the person who underwent 4 times of balloon angioplasty procedures. Multiple palliative operations and catheter interventions were required because of severe diffuse pulmonary arterial hypoplasia, such as right ventricle to pulmonary artery conduit interposition, pulmonary artery angioplasty, and modified B-T shunt operation. He ultimately underwent a Rastelli operation.
The mortality rate of the patients who underwent cardiac operations was 16.7%, and the mean follow-up duration was 4.64±4.89 years (range, 0.02-16.5 years). The preoperative level of serum total bilirubin was similar to the postoperative level observed on postoperative day 30. The mean maximum total bilirubin was 11.45±8.04 mg/dL preoperatively, and 17.90±21.68 mg/dL postoperatively. The comparison of pre- and postoperative total bilirubin levels shows no statistically significant difference (P=0.372). The Aristotle score was used to determine the procedure- adjusted complexity score. The 4 patients had an Aristotle score of ≥9. In 2 of the 4 patients, the level of maximum total bilirubin on postoperative day 30 was ≥15 mg/dL. One of them died on postoperative day 6. We found no statistically significant correlations between the Aristotle score and the level of maximum total bilirubin on postoperative day 30 (P=0.197). However, an Aristotle score of ≥9 with the level of maximum total bilirubin of ≥15 mg/dL on postoperative day 30 was a significant predictive value of mortality on postoperative day 30 (P=0.022).
5. Overall outcome of AGS
Eight patients died at a median age of 4 years (range, 3 months to 15 years). The 5-year survival was 75% with severe liver disease, and 16.7% with combined severities of liver and heart. The causes of death are described in Table 2. Among the 3 patients with severe liver disease, 2 presented with liver failure before the liver transplantation. Hypertrophic cardiomyopathy was found and progressed in one patient who was admitted because of liver failure. One patient who underwent the B-T shunt operation due to pulmonary atresia died suddenly at admission for evaluation of cyanosis.

Causes of death for 8 patients with Alagille syndrome
In the univariate analysis, the survival rate was worse in patients with cardiac anomaly found on fetal ultrasonography than in those with late onset of cardiac presentation (50% at fetal vs. 75.0% at 1-4 months vs. 90.5% at 0-30 days, P=0.009). In 41 patients with AGS, the overall survival was worse in patients with combined severities than in those with severe liver disease or severe heart disease or without severity (28.6% with combined severities vs. 75.0% with severe liver disease vs. 100% with severe heart disease vs. 100% without severity, P<0.001) (Fig. 1).
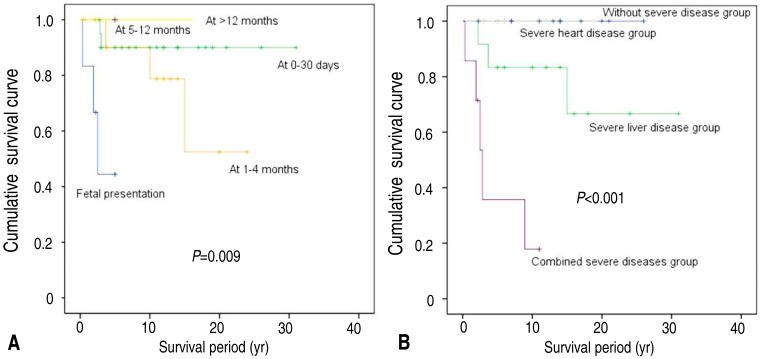
Kaplan-Meier survival curves. (A) The survival rate was worse in patients with a cardiac anomaly that was detected via fetal ultrasonography than in those with later cardiac presentation (50% at fetal ultrasonography vs. 75.0% at 1-4 months vs. 90.5% at 0-30 days vs. 100% at >5 months, P=0.009). (B) Overall survival was worse in patients with combined severe diseases than in those with only severe liver disease or severe heart disease, or in those without severe disease (28.6% with combined severe diseases vs. 75.0% with severe liver disease vs. 100% with severe heart disease vs. 100% without severe disease, P<0.001).
Discussion
AGS is a multisystem syndrome that was first reported in 1969, when the concept of chronic cholestasis in early childhood with heart anomalies as "arteriohepatic dysplasia" was introduced123). Our study states the presentations with onset time, liver evaluation, and cardiac phenotype and intervention. Cardiovascular anomalies were detected using echocardiography or cardiac catheterization in 87.8% of patients. The most common phenotype of cardiac anomalies was pulmonary stenosis, which was present in 97.2% of patients. The most common subtype of peripheral pulmonary stenosis was present in 71.4% of patients. Although the percentage of cardiovascular anomalies in our study was similar to that reported by Alagille et al.12), it was smaller than that reported by Emerick et al.13), which is probably due to the small case size. The severity of the cardiovascular presentation is defined based on the need for intervention or the critical condition, and the cardiovascular presentation is categorized as a right-sided, left-sided, or other lesion. Ten of the 13 patients had a right-sided lesion, 2 had a left-sided lesion, and another patient had hypertrophic cardiomyopathy, which is neither right-sided nor left-sided. Although AGS has been considered as a disease of the right side of the heart, left-sided cardiovascular anomalies were reported in 15% of population. Three patients were diagnosed with tetralogy of Fallot as a right-sided lesion, specifically the severe form tetralogy of Fallot with pulmonary atresia. These results suggest that the patients with AGS who were diagnosed with tetralogy of Fallot had a higher incidence of the severe form with pulmonary atresia than the 20% reported in the general population14). These findings are consistent with McElhinney et al.15). Two of them deceased; 2 (67%) died from cardiovascular causes and the remaining one patient survived. Two patients confirmed the JAG1 mutation, and the other had not performed the chromosomal study because of death before full evaluation. These findings imply that the poor outcome of the patients with tetralogy of Fallot in AGS is notable and the tetralogy of Fallot with pulmonary atresia is needed for additional study about investigation of AGS.
Twelve patients underwent cardiac surgeries. We investigated whether the paucity of hepatic ducts in AGS could be attributed to the outcome of the cardiac operation for the combined cardiac anomalies. Given our finding that the complexity of cardiac operation with an Aristotle score of ≥9 and a maximum total bilirubin level on postoperative day 30 of ≥15 mg/dL was a poor prognostic factor, the presence of higher bilirubin during the postoperative period could be worsen the morbidity/mortality associated with the cardiac operation. It is plausible that the neonatal cholestatic jaundice reported by Lykavieris et al.16) was an independent prognostic factor. However, the correlation between severe liver disease and severe heart disease is not significant. Our study has a limitation because our data treated total bilirubin only. Further studies are needed to investigate about the relationship between conjugated bilirubin and prognosis.
Our study had one patient (2%, n=41) with combined noncardiac structural vascular anomalies. The frequency of vascular abnormalities with AGS was smaller than those reported by Kamath et al.17), which is probably due to the undersized study cases. The patient showed renal artery stenosis leading to intractable hypertension. The gene study was not performed. Although he underwent several trials to resolve the stenosis, the trials failed due to the difficulty of the approach. He is currently on medical treatment with limited effects, and is being considered for surgical treatment (such as nephrectomy or angioplasty).
The mutational analysis identified the JAG1 gene in 28 patients. Twenty-seven of them had 28 JAG1 gene mutations including frameshift, missense, nonsense, splice site, in-frame deletion, and whole/partial gene deletions. The mutations in the NOTCH2 gene are a rare cause of AGS1018). Therefore, it is plausible that a NOTCH2 gene mutation may be present in the remaining patient who met the clinical diagnostic criteria of AGS. Several studies have investigated the JAG1 gene's hotspot and the correlation between genotype and phenotype of AGS. The mutation of the JAG1 gene is through the whole protein19). About 60% of de novo mutations are confirmed2021). These observations including our study reveal that the relationship between the genotype and phenotype of AGS has no apparent correlation1521222324). We therefore suggest that the numerous genetic etiologies, epigenetic factors, and variable environmental modifiers affect the cardiac expressivity of AGS.
Two patients died within 30 postoperative days. One of them was diagnosed with tetralogy of Fallot with pulmonary atresia combined with major aorto-pulmonary collateral arteries (MAPCAs), and underwent 2 operations, namely right ventricular outflow track reconstruction and unifocalization of MAPCAs with angioplasty of the pulmonary arteries. After the second operation, he had extracorporeal membrane oxygenation (ECMO) and died from ECMO weaning failure. Another patient had severe pulmonary stenosis, which presented a combined conflicting risk and benefit with regard to the operation. The liver function progressed to failure and the patient needed liver transplantation. While waiting liver transplantation, the patient underwent pulmonary artery angioplasty operation at 8.83 years of age. She had an emergency decompression operation for intracranial epidural hemorrhage on postoperative day 1, and died of multiorgan failure following brain death. Intracranial bleeding is a major cause of morbidity and mortality1725). Emerick et al.13) reviewed 92 patients with AGS described that 13 patients experienced intracranial bleeding without trauma, and 4 of these patients died. They reported that one patient died from spontaneous fatal intracranial bleeding after the second liver transplantation. We highlight the need for patients with AGS to undergo an evaluation of their intracranial structure before the liver transplantation/cardiac operation.
This study describes the clinical characteristics and the significant prognostic factors of AGS at a single center in Korea. There is a paucity of literature on the outcome with AGS and the correlation of each presentation in Korea. The small sample size in our study gives little power to detect such relationships. However, our observations warrant additional studies with specific presentations and prognostic factors.
In conclusion, AGS must be considered in patients with features that meet the clinical criteria. Surveillance, assessment, and familial investigation should be followed to the cardiologists, hepatologists, and geneticists. The high suspicions based on the clinical manifestations and findings of the genetic evaluation will help to diagnose AGS. The patients with AGS who presented severe cardiovascular anomalies need appropriate surgical or transcatheter interventions, and a multidisciplinary approach is necessary to improve the outcome.
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.

