


Introduction
Neonatal diabetes mellitus (NDM) presenting within the first 6 months of life includes permanent neonatal diabetes mellitus (PNDM), which require lifelong therapy, and transient neonatal diabetes mellitus (TNDM) where the condition shows remission during infancy but relapses in adolescence. Almost all cases of neonatal diabetes have monogenic etiology in contrast to the autoimmune diabetes presenting in children beyond 6 months of age. PNDM is associated with defects in pancreatic beta cell development and function. Activating mutations in the KCNJ11 gene, encoding the subunit Kir6.2, and ABCC8 gene, encoding the sulfonylurea receptor 1 (SUR1) of ATP-sensitive potassium (KATP) channel, which has a key role in insulin secretion in glucose metabolism, are the most common causes and account for approximately 40% of all cases of PNDM1,2). More than 30 mutations in the KCNJ11 gene have been identified2,3). Mutations in the glucokinase (GCK) and insulin (INS) genes have also been reported in patients with PNDM.
Sulfonylureas close the KATP channel by an ATP-independent route, leading to increase insulin secretion4). Therefore, in many patients with PNDM with Kir6.2 and SUR1 mutations, insulin therapy can be replaced by oral sulfonylureas which offer more improvement in glycemic control and better quality of life3,5).
We report a case of PNDM caused by a novel p.H186D heterozygous mutation in the KCNJ11 gene whose treatment was successfully transitioned from insulin to oral sulfonylurea.
Case report
A female child was born at 38-week age of gestation with intrauterine growth retardation. She was delivered by cesarean section with a birth weight of 2.5 kg. She was diagnosed with neonatal diabetes at 2 months of age. She was the first child of two siblings. Her parents were unrelated, and her family was healthy and had no history of diabetes. Laboratory results at diagnosis were: blood glucose, 1,041 mg/dL; pH, 7.025; HCO3-, 5.1 mmol/L; pCO2, 19.8 mmHg; sodium, 147 mmol/L; potassium, 5.6 mmol/L; chloride, 113 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.2 mg/dL; urine ketone, +++; glycosylated hemoglobin (HbA1c), 7.7%; and C-peptide, 0.2 ng/mL. After recovery from diabetic ketoacidosis, she had injections of neutral protamine Hagedorn (NPH) insulin two times a day and regular insulin (RI) four times a day. Her maximal daily insulin dose was at 1.5 U/kg. Her insulin requirement was gradually decreased to 0.44 U/kg/day by 4 months of age. She took NPH and RI injection twice a day from 10 months of age. Although there were several hypoglycemic events, insulin injection for glucose control after the infancy period could not be stopped.
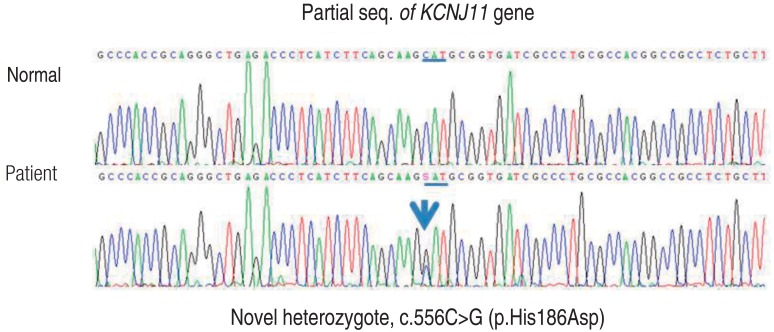
Genetic study for neonatal diabetes using genomic DNA extracted from peripheral lymphocyte was done at 4.5 years of age, and a novel heterozygous mutation of the KCNJ11 gene located on chromosome 11p15.1 and encoding Kir6.2, c.556C>G (p.His186Asp), was found (Fig. 1). Genetic study for her parents could not be done.
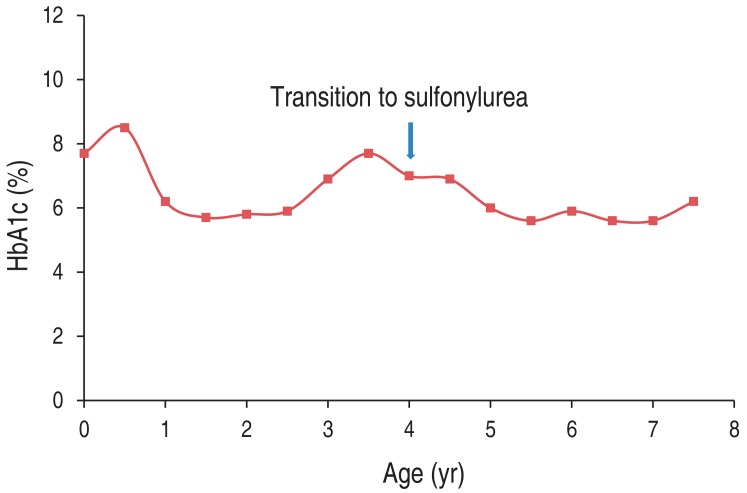
Transition of treatment from insulin to sulfonylurea was attempted at the age of 4.8. Before the trial, her daily insulin requirement was at 0.44 U/kg. Laboratory findings were as follows: HbA1c, 6.9%; fasting glucose, 205 mg/dL; C-peptide, 0.84 ng/mL; 24-hour urine C-peptide, 2.2 ┬Ąg; negative for islet cell antibody; anti-insulin antibody; and anti-GAD antibody. She was 103.2 cm in height (25th-50th percentile), 17 kg in weight (50th-75th percentile) and did not have any specific neurological deficit. Oral sulfonylurea (glibenclamide) was initiated at a dose of 0.1 mg/kg/day divided into two equal doses. The glibenclamide dose was gradually increased to 0.4 mg/kg/day over 1 week and the insulin was stopped. Oral glucose tolerance test (glucose, 1.75 g/kg) was done at 6 months and 1 year after completion of the sulfonylurea transition. The results demonstrate improvement of insulin and C-peptide sercretory response during treatment. Insulinogenic index [╬öInsulin (30 min-0 min)/╬öGlucose (30 min-0 min)], a marker of beta cell function, became increased from 0.10 to 0.18, and acute C-peptide response [╬öC-peptide (30 min-0 min)├Ś1,000/Glucose (30 min-0 min)] became increased from 6.48 to 8.29 (Table 1). The patient is now 7.5 years of age with a height of 121.4 cm (50th percentile) and weight of 23.4 kg (50th percentile). Recent glibenclamide dose is 0.3 mg/kg/day and HbA1c is 6.2%. Blood glucose was well controlled without episodes of hypoglycemia and the HbA1c has been lower than during insulin injection (Fig. 2). No symptoms suspicious for side effects of sulfonylurea have been noticed since.
Discussion
NDM is a monogenic form of diabetes that presents within the first 6 months of life. Heterozygous activating mutations in the KCNJ11 and ABCC8 gene encoding the subunits Kir6.2 and SUR1 of KATP channel in pancreatic beta cells are the most common and account for nearly half of all cases of PNDM1,2). Mutations in the glucokinase (GCK), insulin (INS) genes have also been reported in patients with PNDM. The causes of rare syndromic PNDM includes recessive mutations in several genes, such as PDX1, EIF2AK3, GCK, PTF1A, FOXP3, NEUROG3, NEUROD1, RFX6, IER3IP1, HNF1B, GLIS3, PAX6, SLC19A2, SLC2A2, and WFS16,7). In TNDM, imprinted locus at chromosome 6q24 seen in more than half of cases8), and KCNJ11 and ABCC8 gene mutations are also found in some cases.
Glucose increases intracellular ATP level and it induces the closure of KATP channels that lead to insulin secretion by pancreatic beta cells. Activating mutations in the KCNJ11 or ABCC8 gene of KATP channel lead to KATP channels remaining open despite the presence of glucose, thus, insulin secretion cannot be increased1). On the contrary, loss of function mutations cause congenital hyperinsulinemia due to the closure of the KATP channel and lead to increased insulin secretion.
Sulfonylureas (SU) can close the KATP channel and increase insulin secretion4). Many cases with KCNJ11 and ABCC8 mutation can be treated with oral sulfonylureas. Treatment response is not expected in patients with mutations in other genes, such as glucokinase gene, FOXP3, and IPF1. Therefore, molecular diagnosis in neonatal diabetes may help identify patients that are likely to respond to oral sulfonylureas.
Pearson et al.3) reported the responses to sulfonylurea according to mutation types in diabetic patients caused by KCNJ11 mutation. Switching from insulin to SU was successful in 90% of patients. There were no differences in blood insulin, C-peptide level, and insulin injection dose between the successful group and unsuccessful group. R201H mutation was the most common and all patients responded well to sulfonylurea. V59M, R201C, F35V, H46Y, R50Q, G53R, R201L, E322K, Y330S, F333I mutations also belong to the successful group. Switching to SU was unsuccessful in patients with Q52R, I296L, and L164P mutations. In addition, failure of switching to SU was more related with the presence of neurologic features (14% in successful group and 80% in unsuccessful group) and older age at initiation of SU. Median age was 6 years old (intraquartile range, 3-12 years) in the successful group and 18 years old (intraquartile range, 6-35 years) in the unsuccessful group3). The report about a mother and daughter carrying the same KCNJ11 mutation showed that the daughter could be switched from insulin to SU at age 8.5 years, but her mother had an imcomplete response9). The trial of SU switching in a poorly controlled diabetic 19 years old patient with R201H mutation, a mutation type expected to respond successfully, failed10). These observations suggest that factors that can affect the success of switching to SU include; mutation type, severity of mutation, and duration of the diabetes. A long-standing diabetes may lead to islet beta cell exhaustion resulting in nonresponse to SU9).
NDM caused by KCNJ11 gene mutations is inherited in autosomal dominant pattern, but most of these cases result from new mutations without family history. The patient in this case has a novel mutation (p.H186D) in the KCNJ11 gene and presented with a mild form of PNDM. Switching to SU with an initial dose 0.4 mg/kg/day was successful at 4.8 years of age. Oral glucose tolerance test in index patient showed improvement in insulin secretion during the treatment at 6 months and 12 months after the use of SU. HbA1c level was decreased from 6.7% to 6.2% after SU treatment. Although she had hypoglycemia every 2 or 3 weeks and experienced a few episodes of hypoglycemia with mental change during insulin treatment, there was no more hypoglycemia after changing to SU treatment.
Oral sulfonylurea therapy is both safe and better than insulin for metabolic control. Reported side effects of oral SU are transitory diarrhea and tooth discoloration in a few patients3,11). Lower HbA1c level was observed in SU therapy compared to insulin injection. Mean HbA1c was 8.1% in patients with insulin injection and 6.4% in patients with SU therapy3,12).
Chronic SU treatment seems to develop no beta cell desensitisation in PNDM patients with KCNJ11 mutation13). Early SU therapy at disease onset can permit insulin hypersensitivity and maintained basal insulin secretion, then provide long-term remission in animal subset14). Allowing for the potential beneficial effect on neurodevelopmental outcome and glycemic control, empiric tiral of SU before genetic testing in neonatal diabetes patients can be considered15).
In conclusion, as the mutation type, severity of mutation, and duration of the diabetes are the major factors affecting the success of switching to SU, early genetic analysis and trial of sulfonylurea is important in the management of neonatal diabetes. Further studies looking at the result of SU treatment and long term follow in PNDM patients are needed.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


