


Introduction
Chronic inflammatory demyelinating polyneuropathy (CIDP) is characterized by hyporeflexia or areflexia and progressive or relapsing motor and/or sensory dysfunction of more than one extremity, developing over at least 2 months. While the incidence among adults is 1.0-1.9 per 100,000, the CIDP incidence among patients under age 20 is 0.48 per 100,0001). In addition, because absolute diagnostic tests have not yet been established, CIDP diagnosis in children is difficult to make. However, the biological mechanisms of CIDP children are similar to those of adults, diagnosis criteria and treatment for adults are applied to children2,3,4,5). CIDP diagnostic criteria were developed in 2010 by the European Federation of Neurological Societies/Peripheral Nerve Society Guideline3). The main criteria include (1) progressive or relapsing motor and sensory dysfunction of more than limbs of a peripheral nerve nature, developing over at least 2 months; and (2) electrophysiological evidence of acquired demyelination. The main criteria include (1) progressive or relapsing motor and sensory dysfunction of more than limbs of a peripheral nerve nature, developing over at least 2 months; and (2) electrophysiological evidence of acquired demyelination. Supportive diagnostic tools provide additional important information beyond the main criteria and include cytoalbuminologic dissociation in cerebrospinal fluid (CSF) examinations, magnetic resonance imaging (MRI) findings, objective clinical improvement following immunomodulatory treatment and electron microscopic findings of biopsied nerve3). Here, four patients who were diagnosed with CIDP of acute onset and relapsing courses at Severance Children's Hospital, Seoul, Korea, were enrolled in this retrospective study. These patients were definitely diagnosed by clinical and electrodiagnostic criteria, and treated with intravenous immunoglobulin (IVIG)/corticosteroids or corticosteroids alone.
To understand the clinical courses of CIDP, we report variable CIDP courses in children, with respect to initial presentation, responsiveness to medical treatment, and relapse intervals.
Case report
Patient 1 was an 8-year-old previously healthy girl, who presented gradual symptom of weakness on four extremities 2 weeks after an upper respiratory infection. She could neither use chopsticks nor walk without support. She showed a severely ataxic gait, and her muscle power was subnormal (3/5) in the distal arms and legs. Deep tendon reflexes (DTRs) decreased in the distal extremities. There was nothing remarkable regarding cranial nerve function, and sensory function including touch, pain, vibration, and position were intact. Normal bladder function was maintained. Laboratory tests including complete blood cell count, serum muscle and liver enzyme levels, autoantibody screening, and thyroid function tests were all normal. On CSF examination, the protein level (37 mg/dL) and white blood cell (WBC) count (0 cell/µL) were within the normal range. The initial impression was "probable, acute cerebellar ataxia," based on the post infectious episode, severe ataxia, and normal CSF findings. Brain and spine MRIs were normal. She was given intravenous (IV) dexamethasone (0.3 mg/kg/day) over 3 days, responded rapidly, and was maintained on oral prednisolone (30 mg/day) after discharge. Approximately 20 days later, she presented again with dysarthria, dysphagia, tongue deviation on the right side, and bilateral incomplete eyelid closure for the prior 5 days. The second brain MRI and MR angiography showed no pathologic findings. She was given IV dexamethasone (0.3 mg/kg/day) and responded again within 1 week. She was also given IVIG (400mg/kg/day) for 5 days, and oral prednisolone was continued. Five weeks later, she presented with both arm and leg weakness after 1 day of fever. Pain and touch sensations were impaired on the distal upper extremities. Severe ataxic gait also existed. At the time of the third relapse, an NCS was performed; sensory action potentials in the left ulnar nerve were prolonged and lowered in amplitude. In motor conduction, abnormal findings were noted, with prolonged latency and low amplitude of compound muscle action potential, slow conduction velocity, and temporal dispersion of compound action potentials without definite blocking. These findings suggested demyelinating, mixed sensorimotor polyneuropathy. CIDP was diagnosed by the European Federation of Neurological Societies/Peripheral Nerve Society Guidline3) and she received IV dexamethasone (0.3 mg/kg/day) for 7 days and oral prednisolone (30 mg/day). After 15 days, she had recovered sufficiently to walk and run independently, and the regimen was changed to alternating doses every other day, she recovered in 3 months
Patient 2 was a 10-year-old boy who visited our clinic with a fourth episode of both arm and leg weakness, and difficulty using stairs for 5 days. Though the CSF examination showed no remarkable findings during the first attack, the NCS revealed sensorimotor polyneuropathy, and he was diagnosed with Guillain-Barré syndrome (GBS). However, he relapsed every 3 months after his first episode until fourth episode. On physical examination, muscle power was subnormal (4/5) in both arms and legs. DTRs were reduced in the elbows and knees. Cranial nerve function and sensory function were normal. Laboratory indices, and brain and spine MRI showed no abnormalities. The patient was diagnosed as CIDP with a relapsing course, and received a 5-day regimen of IVIG and IV dexamethasone (0.3 mg/kg/day) followed by oral prednisolone (30 mg/day) for 1 month. He recovered from his symptoms in 2 months.
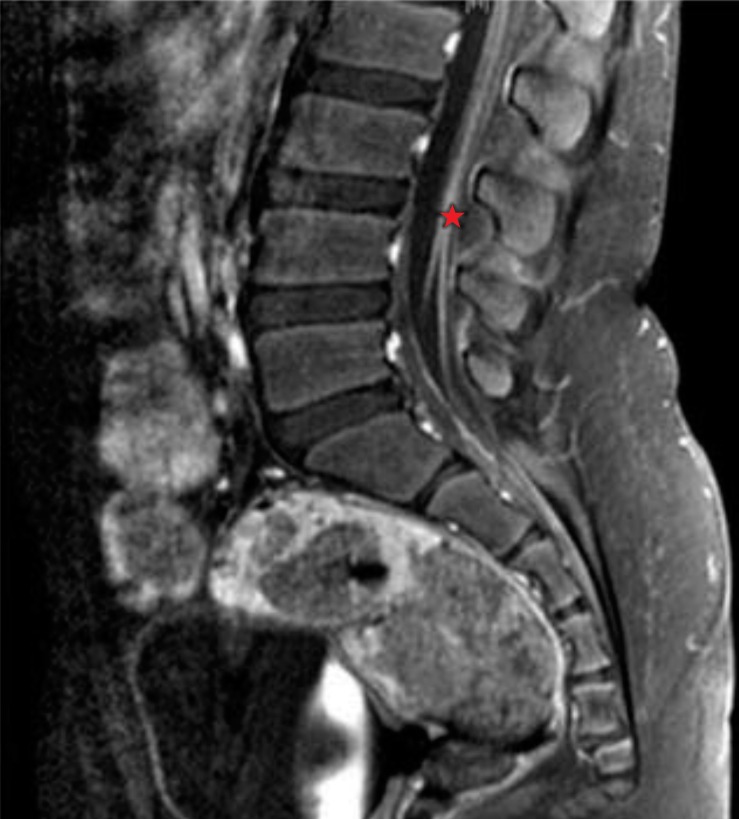
Patient 3 was a 9-year-old boy who had gradual weakness in both lower extremities followed by chest discomfort 1 week after an upper respiratory infection. He was admitted into another hospital, where he received IVIG (400 g/kg/day) for 5 days. He recovered gradually and was able to walk with support after 2 months. Weakness in the lower extremities worsened again 3 months later, prompting him to visit our Emergency Department. He presented with an ataxic gait and required a wheelchair. On physical examination, muscle power was decreased to 4/5 in the upper extremities and 3/5 in the lower extremities. DTR were reduced on both proximal and distal extremities. Sensory function was intact and laboratory indices were noting remarkable. MRI scans of the brain showed no focal lesions, but spinal MRI images showed symmetric smooth pial enhancement of the cauda equina and conus medullaris (Fig. 1). CSF examination showed a raised protein level (209 mg/dL) with a normal WBC count (1 cell/µL). On NCS, sensory action potentials were absent on the left arm and right leg. Prolonged motor conduction, low amplitude of compound muscle action potential, and slow conduction velocity were found in the left arm and right leg. These findings suggested segmental demyelinating, mixed sensorimotor polyneuropathy combined with axonal involvement. We made a diagnosis of CIDP. He was given IVIG (400 mg/kg/day) for 5 days and IV dexamethasone (0.3 mg/kg/day) for 1 week, after which he recovered enough to walk with support. He was maintained on oral prednisolone (30 mg/day for 2 weeks), and then the dose was reduced to 30 mg/day on alternating days for 2 months. He could walk independently more than 300 meters nonstop.
Patient 4 was a 12-year-old girl visited our hospital for the second episode of bilateral lower limb weakness in 6 months. Her initial symptoms occurred 1 week after an upper respiratory tract infection. She had previously been treated with IV dexamethasone (0.3 mg/kg/day) for 5 days. On our physical examination, her muscle power was decreased (4/5) bilaterally in the both proximal and distal leg regions. DTR were reduced on both ankles and knees. Brain and spine MRI scans showed no abnormalities. On CSF examination, the protein level (18 mg/dL) and WBC (0 cell/µL) were within normal limits. On NCS, bilaterally decreased interference of tibial anterioris, external hallucid longus, and external digitorum brevis muscles was observed, and the minimal F-wave latencies were prolonged in the left median and ulnar nerves. The patient was diagnosed with CIDP, received a 5-day course of IVIG (400 mg/kg/day) for 5 days and IV dexamethasone (0.3 mg/kg/day) for 1 week, followed by oral prednisolone (30 mg/day). After 1 month, she recovered sufficiently to walk independently, and the prednisolone dose was reduced to alternating doses of 30 mg/day every other day for 15 days and then tapered to 20 mg/day. After 6 months, weakness in both legs relapsed and she could not walk independently. She was given a 5-day course of IVIG (2 g/kg/day) and IV dexamethasone (0.3 mg/kg/day), and was maintained on oral prednisolone (30 mg/day) for 1 month.
The clinical characteristics of four patients are summarized in Table 1.
Discussion
Diagnosis of CIDP is difficult because of the variation in symptoms and signs and its slowly progressive nature. CIDP most frequently starts insidiously and evolves slowly, either in a slowly progressing or relapsing manner, with partial or complete recovery between recurrences. Children have a more relapsing course than adults1).
Differentiating the GBS from CIDP is difficult because their initial symptoms are similar. Our four patients with acute onset CIDP were initially diagnosed with GBS or cerebellitis. Three patients had a previous upper respiratory infection, and limb weakness developed within 1-2 weeks afterwards. In CIDP children, CSF protein level is increased in about 86% of patients1,4), although it provides important supportive information. Only patient 3 showed albuminocytologic dissociation in the CSF study.
The broadly accepted step for confirmative diagnosis is NCS in sensory and motor nerves on at least four nerves including the proximal upper limb, which shows demyelinating patterns characterized by (1) prolonged distal latencies, (2) reduction of motor conduction velocity, and (3) absent or prolonged F-wave latencies by electrodiagnostic criteria3). These patients were diagnosed by NCS; thus electrophysiologic testing, which usually provides the first evidence of demyelination, and is often the only positive evidence among other laboratory tests, is very important for CIDP diagnosis.
It is recommended to consider retesting if the initial electrodiagnostic test does not meet the diagnostic criteria for CIDP3). The electrical signal patterns are similar in the acute form of this disease and in CIDP6). This makes it more difficult to differentiate between these two diseases in initial testing, although some authors reported a more common sural nerve sparing pattern in acute inflammatory demyelinating polyneuropathy6,7).
Sural nerve biopsy may be considered for patients with atypical clinical presentation where the diagnosis is not obvious4,8,9,10). Nerve biopsy is also useful for assessing other diseases such as vasculitis, sarcoidosis, infections, tumor, or amyloidosis7,11). However, nerve biopsy findings are neither specific nor necessary for diagnosis9).
Childhood CIDP is known to respond effectively to IVIG, corticosteroids, and plasmapheresis and have a more favorable long-term outcome than CIDP in adults1,2). It remains unclear which treatment should be the first-line therapeutic regimen for CIDP. While patients 1 and 4 were given only IV dexamethasone at the first episode (0.3 mg/kg/day), patients 2 and 3 were given a combination of IVIG (2 g/kg) and IV dexamethasone (0.3 mg/kg/day). All patients were maintained with oral prednisolone 30 mg/day, but clinical courses were variable in relapsing intervals and severity. Patient 1 relapsed in as little as 2 weeks to 1 month after IV dexamethasone successfully initially controlled the symptoms, despite maintenance therapy with oral prednisolone.
Although there are limitations of a small number of cases, the efficacy of combination therapy with IVIG and corticosteroids can be considered in our result. As suggested by patient 1 outcome, initial management may affect the frequency and duration of relapses but patient 2 showed a relapse in 6 months. IVIG is frequently used as a first-line therapy by several authors12,13), especially in children compared to adults1,2). IVIG has multiple actions on the immune system that might benefit CIDP: providing anti-idiotypic antibodies, blocking macrophage Fc receptors, inhibiting complement activity, and modulating central B-and T-cell function12). The initial dose is usually 2 g/kg for 2-5 days, followed by 1 g/kg at 1-6 week intervals1,3,7). In the IVIG CIDP Efficacy trial, continued treatment with IVIG every 3 weeks helped to maintain remission12,14). It is not clear for how long the treatment should be continued15). The side effects were generally mild, such as headache, nausea, rash, fever, infection, although some serious adverse events (e.g., pulmonary embolism, compromised renal function) occurred1,2,12). However, these complications are rarely found among children2).
The widely-accepted regimen for corticosteroid use is oral prednisone 1-2 mg/kg daily or on alternate days1). Improvement of symptoms is observed on average between 2 weeks and 2 months14). However, the recommended maintenance periods are not established. Moreover, some clinicians fear for severe systemic side effects of long-term corticosteroid use such as osteopenia, growth retardation, hypertension, and hyperglycemia. Steroid-induced appetite increase and weight gain are major complication for children2). In practice, the maintenance periods differ on a patient-by-patient basis. We experienced several relapses during corticosteroid tapering off or shortly after drug discontinuation. Corticosteroids are not useful in pure motor CIDP patients, who have been reported to worsen with corticosteroid treatment4,12). In our patients, pure motor CIDP were not observed by NCS, and all patients responded effectively to corticosteroid therapy, although with variable durations until response occurred. Other immunomodulatory agents such as cyclosporine, azathioprine, methotrexate, and cyclophosphamide are also used to treat CIDP because of the proposed pathogenesis as an immune-mediated mechanism. Although it has not been determined in children, some reports suggest that these agents are effective in adults with CIDP who experience multiple relapses or fail to respond to IVIG or corticosteroid15). Plasmapheresis can be considered in refractory older children; however, severe treatment risks exist including severe coagulopathy, hypotension, anemia, and electrolyte imbalance2,3).
CIDP in children has a more favorable long-term outcome than in adults1). Improvement of symptoms is observed on average between 2 weeks and 2 months1). Relapsing intervals are also diverse, varying from 2 weeks (patient 1) to 6 months (patient 4) in our series. The treatment algorithm for CIDP in children is not yet clear, and more research is needed to determine the optimal therapeutic strategy.
In conclusion, we present four cases of pediatric CIDP patients who showed various clinical courses and treatment responses.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


