|
|
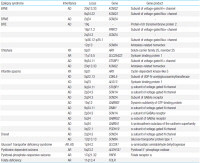
Early-onset epileptic encephalopathies are one of the most severe early onset epilepsies that can lead to progressive psychomotor impairment. These syndromes result from identifiable primary causes, such as structural, neurodegenerative, metabolic, or genetic defects, and an increasing number of novel genetic causes continue to be uncovered. A typical diagnostic approach includes documentation of anamnesis, determination of seizure semiology, electroencephalography, and... |









")

